Krankheitsbilder
Therapie des akuten Schubes
Entstehung von epileptischen Anfällen
Epileptische Anfälle entstehen in der grauen Substanz des Gehirns, meist in der Hirnrinde. Es gibt vielfältige Ursachen, die anfallsauslösend wirken können. Meist liegt ein fehlendes Gleichgewicht zwischen erregenden und hemmenden hirnelektrischen Abläufen vor. Narben, z. B. nach Hirnverletzungen (Geburt, Unfall, Schlaganfall, Hirnentzündungen) oder auch verschiedene Veranlagungen von Zellgruppen (genetische Ursachen, Entwicklungsstörungen) können zu fehlendem Gleichgewicht der hirnelektrischen Vorgänge führen.
Dieses Gleichgewicht zwischen Hemmung und Erregung wird dabei plötzlich in Folge exzessiver lokaler hirnelektrischer Entladungen an solch einem geschädigten Ort durchbrochen. Die epileptische Entladung breitet sich dann aus diesem Herd in die benachbarten Hirnregionen aus. Es kommt zum Ablauf eines epileptischen Anfalls. Herdanfälle sind in ihrer Ausgestaltung vom Ort des Herdes im Gehirn, vom Ausbreitungsweg, von der Intensität und von den hemmenden Mechanismen andererseits des Gehirns und möglicher Medikamente abhängig. Anfälle, die an einem solchen epileptischen Ort entstehen, werden Herdanfälle genannt.
Es kann aber auch ohne nachzuweisende Schädigung des Gehirns zu epileptischen Anfällen kommen. Diese entstehen häufig durch eine zu starke Synchronisierung von elektrischer Hirnaktivität, es kommt zu einem „Aufschaukeln“ hirnelektrischer Vorgänge und ab einem bestimmten Überschreiten von Hemmmechanismen schließlich zu einem generalisierten Ausbruch von epileptischer Aktivität durch das Aufschaukeln selber. Bei den generalisierten Epilepsien laufen solche Ausbrüche epileptischer Aktivität gleichzeitig über mehrere Hirnstrecken ab. Solche Anfallsursachen spielen häufig bei vererbbaren Epilepsien eine Rolle und insbesondere auch bei den altersgebundenen Epilepsien, also bei den Epilepsien des Kindes- und Jugendalters. Hier kommt es häufig im Erwachsenenalter zum Verschwinden der Anfälle.
Diagnose
Nicht jede Bewusstseinsstörung oder verkrampfende Muskulatur bedeutet, dass ein epileptischer Anfall abgelaufen ist. Es kann auch beim Kreislaufkollaps (Synkope) zu Bewusstseinsverlust und sogar auch zu einer Verkrampfung der Muskulatur kommen.
Auch gibt es Bewusstseinsveränderungen, die wie ein epileptischer Anfall anmuten, aber andere Ursachen haben, wie z. B. psychogene (dissoziative) nichtepileptische Anfälle und Affektkrämpfe im Kindesalter. Ein nur einmalig erlebter zerebraler Anfall kann möglicherweise auch nur provoziert sein und lässt nicht unmittelbar die Diagnose einer Epilepsie zu. Wenn ein unprovozierter oder mehrere nicht provozierte Anfälle abgelaufen sind und sich auch im EEG (Hirnstromaufzeichnung, Elektroenzephalogramm) typische epilepsierelevante Veränderungen finden, ist von der Diagnose eines zerebralen Anfallsleidens (Epilepsie) die Rede. Zerebrale Anfälle können als Begleitsymptome von Hirnerkrankungen (Schlaganfall, Hirntumor, Hirnnarben, Stoffwechselerkrankungen) auftreten.
Grundlagen für Anfallsabläufe
Entspringt der Anfall einem bestimmten Ort im Gehirn (Herdanfall) richten sich die Symptome und die Ausgestaltung des Anfallablaufs nach dem Ort der Entstehung und der Ausbreitung im Gehirn. Bestimmte Hirnareale sind für bestimmte Hirnfunktionen zuständig. Im zerebralen Anfall kommt es dann zu einer meist auf den Ort der Ausbreitung bezogenen Veränderung des Verhaltens bzw. Veränderung der hier lokalisierten Hirnfunktionen.
Die Herdanfälle sind deshalb häufig bei verschiedenen Patienten nicht identisch und Patienten mit einer herdgebundenen Epilepsie unterscheiden sich in der Ausgestaltung (Semiologie) eines solchen Anfalls.
Die häufigste Form des Herdanfalls ist der Temporallappenanfall bzw. die Temporallappen-Epilepsie. Bei der Temporallappen-Epilepsie kommt es zu psychomotorischen Anfällen. Die psychomotorischen Anfälle gehen mit einer Bewusstseinsstörung einher, ohne dass die Betroffenen das Bewusstsein verlieren müssen. Es kommt auch zu Veränderungen der Bewegungen (Motorik). So durchlaufen die Patienten bestimmte Bewegungsmuster oder sogenannte motorische Schablonen, ohne dass diese willkürlich verändert werden. Die Beeinflussung von außen durch Zurufen oder Schütteln oder Ähnliches ist meist nur eingeschränkt oder gar nicht möglich. Ist das Bewusstsein während des Anfalls verändert, wird der Begriff „komplex“ dafür verwendet. Da ein solcher Anfall aus einem Herd (Fokus) entspringt, benennt man diese Gruppe von Anfällen auch komplex-fokale Anfälle.
Die Anfälle aus einem zur Schädelmitte gerichteten Herd im Temporallappen sind meist komplex-fokale bzw. psychomotorische Anfälle – sie bilden die häufigste Untergruppe.
Verlauf von psychomotorischen Anfällen
Der Ablauf beginnt häufig mit einer Aura. Der Betroffene bemerkt eine von der Magengegend aufsteigende Übelkeit oder ein aufsteigendes Gefühl des Unwohlseins oder Beklemmtheit. Es kann auch Angst, Panik oder Ähnliches hinzukommen; auch Glücksgefühle können bestehen. Bekannt sind auch Halluzinationen von Gerüchen oder Veränderungen der Geruchswahrnehmung während der Aura. Die Aura kann nur Momente bis sekundenlang dauern und entspricht dem Beginn der epileptischen Entladungen in dem Anfallsherd. Mit Ausbreitung der epileptischen Entladungen über benachbarte und weitere Hirnregionen des Herdes verschwindet die Aura. Nun ist der Betroffene meist nicht mehr bei vollem Bewusstsein. Er reagiert nicht auf Ansprache, ist aber wach, und es kommt häufig zu einer kurzdauernden Bewegungsstarre. Daran schließt sich dann ein Nesteln, Lachen, Drehen, Beinausstrecken, Schmatzen an. Häufig verharrt der Patient auch lediglich in der gerade durchgeführten Bewegung. Auch Schlucken, Schmatzen, Lecken und Rülpsen kommen vor. Diese Anfallsymptome dauern insgesamt bis zu 2 Minuten, meist sind sie kürzer. Der Anfall endet prompt. Der Betroffene beginnt sich zu reorientieren und kann sich meist nicht an den Anfallsablauf erinnern; die Aura dagegen kann erinnert werden.
Verlauf anderer Herdepilepsien
Abhängig von dem Ort des Epilepsieherdes variiert die Ausgestaltung der Anfallsabläufe. So gibt es insbesondere bei Herden im Frontallappen auch Abläufe von kompliziertesten motorischen und gestischen Automatismen, bei Anfällen des occipitalen Hirnlappens Störungen des Sehens und der visuellen Wahrnehmung. Bei Temporallappen-Epilepsie mit Ursprung oder Verteilung im Bereich der Sprachregionen können Sprachstörungen im Laufe des Anfalls und nach dem Anfall auftreten.
Verlauf sekundär generalisierter Anfälle
Nicht selten können die aus einem Herd entsprungenen psychomotorischen Anfälle in einen sogenannten „großen Anfall“ einmünden. Solche sekundär generalisierten Anfälle beginnen dann ebenfalls mit einer Aura, gehen in eine Bewegungsstarre und schließlich in eine Muskelstarre über, die tonischer Ablauf genannt wird. Schließlich kommt es zu einem rhythmischen Zucken und Schlagen der Arme und Beine, das den klonischen Teil des Anfallsablaufs darstellt. Solche aus einem Herd sekundär generalisierten tonisch- klonischen Anfälle sind schwerer und ergreifen in der hirnelektrischen Ausbreitung größere und weitere Anteile des gesamten Gehirns. Im Anfallsverlauf kommt es zu einem Zungenbiss oder Einnässen. Der tonisch-klonische Anfall bricht meist nach bis zu 2 Minuten Dauer abrupt ab, und es setzt der Terminalschlaf ein, aus dem der Betroffene nur schwer und nicht vollständig weckbar ist. Auch können nach einem solchen sekundär generalisierten tonisch-klonischen Anfall Hirnfunktionsstörungen festgestellt werden. Sie stammen von Hirnarealen, in denen die epileptischen Entladungen besonders heftig und lang andauernd abgelaufen waren.
Generalisierte Anfälle
Tonisch-klonische Abläufe von zerebralen Anfällen können aber auch ohne Vorläufersymptomatik abrupt einsetzen, z. B. bei generalisierter Epilepsie, wenn es keinen einzelnen Anfallsherd gibt. Zu den generalisierten Epilepsien zählen auch viele der altersgebundenen Epilepsien. Eine häufige altersgebundene Epilepsie ist die Epilepsie des Schulkindalters, auch Absencen-Epilepsie genannt.
Absencen Epilepsie
Bei der Absencen-Epilepsie kommt es üblicherweise nicht zu starken Bewegungsveränderungen, vielmehr starren die häufig 6 – 8jährigen Schulkinder für etwa 10 Sekunden in den Raum oder auf einen bestimmten Gegenstand und sind während dieser Zeit nicht ansprechbar. Sie können sich an diese Bewusstseinslücke nicht erinnern. Nach einer solchen Absence bestehen orientierend keine wesentlichen Veränderungen, und ein solches Kind kann häufig, ohne dass irgendjemand etwas bemerkt hätte, unverändert am Unterricht teilnehmen oder Ähnliches. Solche Kinder können einmal stolpern oder fallen, ohne dass eine Ursache erkannt wird, oder aber mit dem Fahrrad stürzen ohne eine von außen erkennbare Ursache. Ursache ist vielmehr die Absence.
Psychogene (dissoziative) nichtepileptische Anfälle
Die Differentialdiagnose psychogener Anfällen versus epileptische Anfälle ist schwierig. Häufig werden psychogene epileptische Anfälle insbesondere bei Therapieresistenz nicht erkannt. Im Mittel sollen bis zu 7 Jahren Krankheitsdauer vergehen, bis es zur sicheren Diagnose von psychogenen Anfällen kommt.
Psychogene nichtepileptische Anfälle sind Ereignisse, die epileptischen Anfällen ähneln, allerdings durch psychische Prozesse ausgelöst sind. Sie sind nicht durch epileptische neuronale Störungen verursacht.
Die Diagnose lässt sich meist durch Beobachtung der Anfälle stellen. So ist bei psychogenen nichtepileptischen Anfällen die Anfallsdauer allgemein länger als 2 Minuten, und es kommt auch durchweg zum Lidschluss bei psychogenen nichtepileptischen Anfällen. Bei epileptischen Anfällen ist beides sehr selten und ein epileptischer Anfall dauert nur selten länger als 2 Minuten. Bei psychogenen nichtepileptischen Anfällen bestehen häufig eine psychiatrischen Behandlung, häufig multiple Operationen und invasive Untersuchungen sowie häufig multiple unerklärte körperliche Beschwerden. Auch sexueller und physischer Missbrauch ist häufig. Diese genannten Auffälligkeiten sind bei epileptischen Anfällen selten.
Ein drei- bis fünffaches Ansteigen des Basalwertes von Prolaktin oder Kortisol im Blutserum etwa 15 – 20 Minuten nach dem Anfall ist bei etwa einem Drittel der epileptischen Anfälle feststellbar. Hormonanstiege bei psychogenen Anfällen können aber auch vorkommen.
Im EEG lässt sich bei psychogenen Anfällen keine epileptische Aktivität nachweisen. Häufig kann zur Diagnose auch eine längere Video-EEG-Beobachtung beitragen. Oft lassen sich auch bei psychogenen Anfällen Anfälle durch Gabe eines Placebos provozieren und beenden. Es kann auch zu Kombinationen von „echten“ epileptischen Anfällen und zusätzlichen psychogenen nichtepileptischen Anfällen kommen, was die Diagnostik erschwert.
Unterscheidung zwischen epileptischem und psychogenen Anfall
nach Reuber et al. 2003
Die Therapie muss auf die jeweiligen einzelnen Patienten abgestimmt werden, da die Verursachung nicht einheitlich ist. Hier muss insbesondere bedacht bleiben, dass auch sexueller und physischer Missbrauch zu psychogenen Anfällen führt. Diagnostische Zuordnung insbesondere zu posttraumatischen Belastungsstörungen und Angststörungen sind sinnvoll. Psychotherapie und Soziotherapie sollten eingesetzt werden. Auch die Einnahme von Serotonin-Wiederaufnahmehemmer kann positiv sein.
Selbst bei Diagnosestellung können die Patienten nur schwierig ihr Krankheitsbild akzeptieren und bestehen auf Fortfahren der antikonvulsiven Medikation obwohl keine Epilepsie vorliegt. Behandlungen in psychosomatisch orientierten Kliniken, die sich insbesondere mit dissoziativen Störungen befassen, sind sinnvoll.
- Reuber, M. und Bauer, J.: Psychogene nichtepileptische Anfälle. Deutsches Ärzteblatt Heft 30, Jahrgang 100, S. 2013 – 2018, 2003.
- Reuber, M., Fernandez, G., Bauer, J. et al.: Diagnostic delay in patients with psychogenic non-epileptic seizures. Neurology 58: 493 – 495, 2002.
- Stefan, H.: Epilepsien. Diagnose und Behandlung. Georg Thieme Verlag 1999, Stuttgart.
Verbreitung der Krankheit
Die jährliche Rate an Neuerkrankung für Epilepsie wird in entwickelten Ländern mit etwa 40 – 70 Erkrankungen auf 100.000 Einwohner angenommen (Inzidenz). Weltweit wird geschätzt, dass etwa 3 – 5% der Bevölkerung im Laufe ihres Lebens an einer Epilepsie erkranken (Prävalenz), dies allerdings nur vorübergehend. Epilepsie ist eine der häufigsten chronischen Erkrankungen im neurologischen Fachgebiet. Die Häufigkeit aktiver Epilepsien zu einem bestimmten Zeitpunkt wird auf 0,5 – 1 % der Bevölkerung geschätzt. In Deutschland leben etwa 500.000 Menschen mit behandelter aktiver Epilepsie. Bei Menschen mit geistiger Behinderung beträgt die Prävalenz 20 %. Dies liegt daran, dass Hirnschädigungen häufig auch Epilepsien verursachen. Man schätzt, dass etwa 400.000 Patienten in Deutschland an Epilepsie behandelt werden. Von diesen 400.000 Patienten werden etwa 120.000 von den Hausärzten allein behandelt. Hausärzte überweisen ca. 75 % dieser Patienten an Neurologen oder Nervenärzte. Besonders Patienten mit fortbestehenden Anfällen oder Nebenwirkungen der Medikamente werden an Epilepsie-Ambulanzen weitergeleitet (ca. 7 – 10 %). Mehr als zwei Drittel der Patienten sehen durch die Epilepsie Beeinträchtigungen im täglichen Leben. Der Anteil der erwerbstätigen Epilepsie-Patienten beträgt nur 50 % der allgemeinen Erwerbstätigkeitsquote.
Therapie
Die Medikamente gegen Epilepsie (Antiepileptika) wirken über eine Verstärkung der hemmenden hirnelektrischen Vorgänge, oder aber sie verhindern das Aufschaukeln bzw. die rhythmische überschwellige Entladung der Nervenzellen. Erst wenn die Diagnose von zerebralen Anfällen (epileptische Anfälle) durch den Neurologen oder Nervenarzt gesichert ist, sollten diese Medikamente eingenommen werden. In fast 60 – 80 % der Fälle tritt mit guter medikamentöser Einstellung kein Anfall mehr auf. Patienten, die mit Medikamenten aber weiterhin Anfälle haben, können in speziellen Epilepsie-Zentren untersucht und auch operiert werden, so dass durch die Operation Anfallsfreiheit eintritt.
Therapieziel ist die Anfallsfreiheit. Es gibt zahlreiche bewährte, seit langem eingeführte Antiepileptika und zahlreiche in den letzten Jahren zugelassene neue. Antiepileptika können auch falsch verordnet werden. Beispielsweise gibt es Substanzen, die nur gegen generalisierte Epilepsie helfen, nicht aber gegen Herdanfälle. Nebenwirkungen bestehen bei fast allen Antiepileptika, sie sind häufig gut kontrollierbar. Die Alltagsbefindlichkeit kann durch Nebenwirkungen beeinträchtigt werden; meist wiegt die Beeinträchtigung aber weniger als die Beeinträchtigung durch nicht behandelte Anfälle.
Notwendigkeit der medikamentösen Behandlung
Nach zwei unprovozierten Anfällen sollte nach vorhergehender intensiver neurologischer Abklärung mit Antiepileptika therapiert werden. Kam es lediglich zu einem Gelegenheitsanfall, provoziert durch Schlafentzug, Alkohol, Fieber oder Medikamente, kann auch Lebensregulierung und Verlaufsbeobachtung zunächst durchgeführt werden. Nach Patientenwunsch kann aber bereits ein erster unprovozierter Anfall medikamentös eingestellt werden.
Die Auswahl der Antiepileptika richtet sich nach dem Anfallstyp und erfolgt zuerst nur in Therapie mit einer Medikamentensubstanz (Monotherapie). Sprechen die Anfälle auf das Medikament nicht an, erfolgt die Umstellung auf ein weiteres Medikament der I. Wahl. Bei weiterem fehlenden Ansprechen erfolgt die Kombinationstherapie – häufig auch mit einem der neueren Antiepileptika. Bestehen weiterhin Anfälle und ist die Epilepsie medikamentös nicht anfallsfrei einzustellen, kann die prächirurgische Abklärung erfolgen. Viele therapieresistente Fokalepilepsien sind durch operative Behandlung heilbar oder zumindest anfallsfrei einzustellen.
Bei den primär-generalisierten Epilepsien ist eine Operation selten angebracht. Sie kann sinnvoll sein bei Sturzanfällen (Kallosotomie mit Durchtrennung des Balkens zwischen linker und rechter Hirnhälfte). Auch Elektrostimulation des Vagusnervs kann hier hilfreich sein.
Wer behandelt Patienten mit Epilepsie?
Hausarzt
Kommt es zu Störungen des Bewusstseins oder auch zu abnormen Bewegungsabläufen mit Störung des Bewusstseins oder zu einem tonisch-klonischen Anfall, sollte der Patient sich zunächst an seinen Hausarzt wenden. Dieser kann die allgemeinen Faktoren zur Anfallsauslösung eingrenzen und auch Bewusstseinsveränderungen auf Grund anderer Erkrankungen abwägen. Über Epilepsie des Kindes – Jugendalters wissen die Kinderärzte viel, häufig sind diese auch auf Epilepsien spezialisiert.
Neurologe (Nervenarzt)
Um die näheren Ursachen des Anfalls zu untersuchen, sollte der Patient dann aber an einen Neurologen oder Nervenarzt weitergeleitet werden. Dieser untersucht mit dem Elektroenzephalogramm (EEG) die Hirnströme. Er führt an Hand der Anfallsbeschreibung und Ausgestaltung des Anfalls eine Typisierung des Anfalls durch. Da die Anfallstypisierung zur weiteren medikamentösen Therapie wichtig ist, sollte die Durchführung der medikamentösen Therapie von fachärztlicher Seite (Neurologe) gesichert werden. Bei einem Erstanfall muss nach auslösenden Faktoren der Epilepsie gefahndet werden. Anfälle können auch durch Hirnblutung, Entzündungen des Gehirns, Hirntumoren, Stoffwechselveränderungen ausgelöst werden. Neben der neurologischen Untersuchung ist die Durchführung des Elektroenzephalogramms, eines Kernspintomogramms oder Computertomogramms sowie Laborwertuntersuchungen und gegebenenfalls auch Nervenwasseruntersuchung (Liquor) notwendig. Auch Vergiftungen können zu Anfällen führen.
Kontrolle von Nebenwirkungen
Die medikamentöse Therapie muss wegen der Nebenwirkungen auf das Blutbild, die Leber- und Nierenfunktionen sowie anderer wichtiger Körperfunktionen ärztlich kontrolliert sein. Hier sollten Neurologe und Hausarzt zusammenarbeiten. Von den meisten Antiepileptika lassen sich auch die Konzentrationen des Medikaments im Blut messen. Die Bestimmung dieser Wirkspiegel ist zur Verbesserung der Therapie wichtig; auch können dadurch Überdosierungen vermieden werden. Wichtigstes Ziel bei der medikamentösen Therapie ist die Anfallsfreiheit soweit dies möglich ist. Auffälligkeiten im EEG verbessern sich durch Antiepileptika, es ist aber nicht zwingend bei erreichter Anfallsfreiheit noch vorhandene EEG-Auffälligkeiten zusätzlich medikamentös zu behandeln.
Epilepsie und Schwangerschaft (Update 2020)
Insgesamt stehen über 20 verschiedene Medikamente (Antikonvulsiva) zur Therapie
eines Krampfleidens zur Verfügung. Das primäre Ziel der Therapie ist immer
Anfallsfreiheit bei guter Verträglichkeit der Medikamente.
Bei Frauen bis zum Einsetzen der Menopause gibt es darüber hinaus zu beachten, dass
sowohl ein Krampfleiden, als auch die antikonvulsive Medikation negative Auswirkungen
auf eine Schwangerschaft und die kindliche Entwicklung haben können. Auch
bestehen Wechselwirkungen zwischen Antikonvulsiva und Kontrazeptiva, sodass durch
gegenseitige Enzyminduktion sowohl Antikonvulsiva, als auch Kontrazeptiva weniger
wirksam sein können.
Kein einziges Antikonvulsivum ist laut Hersteller direkt für die Therapie von Schwangeren
gedacht oder zugelassen, aber aus klinischen Studien ist bekannt, welche Medikamente
potentiell gefährlich sind und bei welchen nicht mit Problemen zu rechnen ist.
Deshalb sollte, sofern nötig, vor einer SSW auf ein geeignetes Medikament umgestellt
werden.
Bei Patientinnen mit Epilepsie hat eine geplante Schwangerschaft viele Vorteile. Die
Medikamente und ihre Dosierung kann den Bedürfnissen während Schwangerschaft und
Geburt angepasst werden.
Bei bereits eingetretener Schwangerschaft sollte die Therapie in der Regel nicht mehr
umgestellt werden, um keine Rezidive bzw. eine Verschlechterung der Anfallssituation zu
riskieren. Die Organanlage ist mit der 12. Schwangerschaftswoche weitgehend
abgeschlossen, sodass eine teratogene (fruchschädigende) Wirkung von Medikamenten
vor allem in der Frühschwangerschaft problematisch ist. Die Einnahme von Folsäure kann
protektiv wirken.
Grundsätzlich ist jedoch das kindliche Risiko bei einem epileptischen Anfall der
Mutter höher als das Risiko für eine medikamenteninduzierte Schädigung, sodass
eine antikonvulsive Medikation auf keinen Fall einfach abgesetzt werden sollte.
Ca 2/3 aller Patientinnen sind während der Schwangerschaft anfallsfrei, bei nur 16% findet
sich eine Verschlechterung (Battino et al., 2013; La Neve et al., 2014).
Eine Zunahme an epileptischen Anfällen im 2. oder 3. Trimenon kann auf eine erhöhte
Medikamentenausscheidung und folgend eine erniedrigte Serumkonzentration
zurückgeführt werden, sodass regelmäßige Kontrollen der Medikamentenspiegel
notwendig sind.
Trotz eines Übertritts geringer Mengen an Antikonvulsiva in die Muttermilch, ist Stillen
ohne negative Folgen für das Kind. Der positive Effekt des Stillens ist auch bei Müttern
unter antiepileptischer Therapie belegt (Veiby et al., 2013).
Ein Kaiserschnitt aus Angst vor Anfällen ist nicht indiziert, das Kind kann auf natürlichem
Weg geboren werden. Die Antikonvulsiva sollten wie üblich eingenommen werden.
Copyright © 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Epilepsie und Fahrtauglichkeit (Update 2020)
„Nach §2, Abs. 4 Straßenverkehrsgesetz (StVG), ist zum Führen von Kraftfahrzeugen
(nur) geeignet, wer die notwendigen körperlichen und geistigen Anforderungen erfüllt
und nicht erheblich oder nicht wiederholt gegen verkehrsrechtliche Vorschriften oder
gegen Strafgesetze verstoßen hat.“ (Positionspaper DGN Fahrtauglichkeit)
Epileptische Anfälle am Steuer können zu schweren Unfällen führen. Deswegen gilt
grundsätzlich: Solange ein erhöhtes Risiko für einen epileptischen Anfall besteht, besteht
keine Fahrtauglichkeit. Menschen mit Epilepsie dürfen also nur ein Kraftfahrzeug steuern,
wenn sie anfallsfrei sind.
Wer nach § 315c des Strafgesetzbuches im Straßenverkehr ein Fahrzeug führt, „obwohl er
infolge geistiger oder körperlicher Mängel nicht in der Lage ist, das Fahrzeug sicher zu
führen […] und dadurch Leib oder Leben eines anderen Menschen oder fremde Sachen
von bedeutendem Wert gefährdet, wird mit Freiheitsstrafe bis zu fünf Jahren oder mit
Geldstrafe bestraft.“ (StGB)
Zusätzlich kann es versicherungs- und privatrechtliche Konsequenzen geben.
Es erfolgt eine Unterteilung in Gruppe 1 (PKW; Führerscheinklassen A, A1, A2, B, BE, AM,
L, T) und Gruppe 2 (LKW/Bus; Klassen C, C1, CE, C1E, D, D1, DE, D1E, FzF)
Gruppe 1: Patienten dürfen unter folgenden Bedingungen wieder ein Kraftfahrzeug
führen:
1) Bei einem einmaligen Anfall
- nach einer anfallsfreien Zeit von 3 Monaten bei einem provozierten Anfall
(u.a. Fieber, Schlafentzug, Alkohol) ohne sonstige Hinweise auf erhöhe
Anfallsbereitschaft und Wegfallen der anfallsauslösenden Bedingungen - nach einer anfallsfreien Zeit von 6 Monaten bei einem unprovozierten Anfall
ohne sonstige Hinweise auf erhöhe Anfallsbereitschaft
2) Bei der Diagnose Epilepsie
- nach einer anfallsfreien Zeit von 1 Jahr
- bei ausschließlich an den Schlaf gebundenen Anfällen nach 3-jähriger
Beobachtungszeit - bei einfachen fokalen Anfällen ohne Bewusstseinsstörung oder sonstige
Behinderung nach 1-jähriger Beobachtungszeit - bei einem Anfall nach langjähriger Anfallsfreiheit nach einer Zeit von 6
Monaten
3) Bei der Beendigung einer antikonvulsiven Therapie
- Fahrpause während der Reduzierung der Medikament sowie für die ersten
3 Monate nach Absetzen der Medikation
Gruppe 2: Die Auflagen sind erheblich strenger. Nur unter folgenden Bedingungen sind
Epilepsie-Patienten für das Führen eines LKW und den beruflichen Personentransport
geeignet:
1) Bei einem einmaligen Anfall
- nach einer anfallsfreien Zeit von 6 Monaten bei einem provozierten Anfall
ohne sonstige Hinweise für erhöhe Anfallsbereitschaft und Wegfallen der
anfallsauslösenden Bedingungen - nach einer anfallsfreien Zeit von 2 Jahren bei einem unprovozierten Anfall
ohne sonstige Hinweise für erhöhte Anfallsbereitschaft
2) Bei der Diagnose Epilepsie:
- nach einer anfallsfreien Zeit von 5 Jahren ohne medikamentöse Therapie
(praktisch ist bei einer Epilepsie nicht damit zu rechnen, dass jemals wieder
ein LKW oder Bus gefahren werden kann).
Neuerwerb des Führerscheins: Im Antragsformular der Straßenverkehrsbehörde muss die
Frage nach Vorliegen einer Epilepsie oder chron. Krankheit bejaht werden. Daraufhin
muss i.d.R. ein ärztliches Attest oder ein verkehrsmedizinisches Gutachten vorgelegt
werden, das eine Aussage über die Fahrtauglichkeit trifft.
Für weitere Information/Quelle:
Begutachtungsleitlinien zur Kraftfahreignung – BASt 2017
Positionspaper DGN Fahrtauglichkeit
Holger Grehl, Frank Reinhardt: Checkliste Neurologie, Georg Thieme Verlag, Stuttgart
2016
A. Hufschmidt, C.H. Lücking, S. Rauer: Neurologie compact, Georg Thieme Verlag,
Stuttgart 2013
Copyright © 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Literatur für Patienten
- Schmidt, Dieter: Epilepsienfragen und -antworten, 5. Aufl., Zuckschwerdt-Verlag, 1999
ISBN 3-88603-679-0 - Schmidt, Dieter: Taschenatlas Epilepsien, 1992
ISBN 3-88603-446-1 - Degen, Rolf: Die zerebralen Anfallsleiden – Epilepsien, 1988
ISBN 3-528-07981-9 - Epilepsiebericht 1998, Hrsg. Epilepsiekuratorium,
ISBN 3-9805386-3-X - Heinen, Gerd: Bei Tim wird alles anders. Berlin. Verlag einfälle, 1996
ISBN 3-9805386-0-5 - Informationstafeln Epilepsie (Altrup, Specht. Hrsg), 1997 Novartis Pharma Verlag, 90327 Nürnberg
ISBN 3-929126-09-5
Epilepsie oder Ohnmacht (Synkopen) bei älteren Patienten
Die Häufigkeit von cerebralen Anfällen bzw. epileptischen Anfällen ist bei Patienten, die älter als 65 Jahre sind, höher als bei Kindern und Jugendlichen. Die häufigste Ursache von Epilepsien im höheren Lebensalter sind abgelaufene Durchblutungsstörungen oder Schlaganfälle.
Zum Schutz vor Anfällen können die bekannten Antiepileptika genommen werden. Bei älteren Patienten ist allerdings durch Begleiterkrankungen von internistischer Seite, z. B. der Leber, der Nieren und auch durch zusätzliche Medikamentengabe für mögliche Herzerkrankungen oder andere Erkrankungen die Wahl der Medikamente und ihre Dosierung schwieriger.
Die Verträglichkeit der Medikamente ist im Alter weniger gut. Die Antiepileptika werden deshalb häufig auch ohne Rücksprache des Patienten mit dem Arzt abgesetzt. Hier kommt es auf eine gute Zusammenarbeit zwischen Patient, Angehörigen und Arzt an. Bei älteren Epilepsie-Patienten muss die Dosisfindung sehr sorgfältig erfolgen. Die Aufdosierung erfolgt in der Regel langsam, d.h. in Tagen und Wochen. In etwa 70% aller Fälle kann eine medikamentöse Beherrschung der Anfälle erreicht werden.
Diagnose
Zu Ohnmachten, Stürzen, Dämmerzuständen, Episoden mit Gedächtnisstörungen und ähnlichen Beschwerden, wie sie auch bei cerebralen Anfällen (Epilepsie) auftreten, kommt es im Alter gehäuft. Eine diagnostische Zuordnung des Beschwerdebildes ist vor einer Therapie notwendig und sollte vom Neurologen und Internisten durchgeführt werden.
Dazu benötigt der Arzt zunächst eine exakte Beschwerdeschilderung des Ablaufs der Bewusstseinsveränderung. Dies kann, soweit vom Patienten erinnerlich, vom Patienten selber durchgeführt werden. Besonders wichtig ist aber die Verhaltensdarstellung durch Angehörige oder Beobachtende, da dadurch der Arzt relevante Informationen zur Diagnostik erhält. Beispielsweise lassen sich dadurch bereits wichtige Unterschiede zwischen Ohnmachtsanfällen und epileptischen Anfällen feststellen. Hierzu siehe bitte Tabelle.
Unterscheidung zwischen Synkope und epiletischem Anfall
| Synkope | Epileptischer Anfall | |
|---|---|---|
| Alter | jedes Alter | jedes Alter |
| Provokation | emotionale Belastung | Schlafentzug, spezifische Trigger bei Reflexepilepsien |
| Position | aufrecht | jede Position |
| Tageszeit | Wachen | Wachen und Schlaf |
| Hautkolorit | blass | normal oder zyanotisch |
| Aura | Schwindel, Sehstörung, Übelkeit | vielgestaltig, z.T. ähnlich der Synkope geschildert |
| Beginn | schleichend, selten abrupt | plötzlich oder nach Aura |
| Motorische Symptome | bei Asystolie >15 Sekunden Tonisierung möglich | häufig |
| Zungenbiss/Einnässen | möglich | möglich, bei Grand mal häufig |
| Autonome Störungen | üblich | selten |
| Dauer | kurz | kurz bis länger |
| Verletzung | selten | möglich |
| Einnässen | selten | häufiger vorkommend |
| Postiktale Desorientiertheit | selten, eher rasche Erholung | möglich, langsamere Erholung durchaus zu erwarten |
| Motorische Entäußerung | je nach Dauer Tonisierung | häufig |
| Automatismen | nicht zur erwarten | häufig bei fokalen Anfällen oder Grand mal |
| EEG | interiktal unauffällig, iktal verlangsamt | interiktal häufig abnorme Befunde, iktal Rhythmisierung fokal oder generalisiert je nach Anfallstyp |
Der Neurologe wird zur Überprüfung der Hirnfunktionen ein EEG (Elektroenzephalogramm) und zur Kontrolle der Durchblutungssituation eine Ultraschalluntersuchung der gehirnversorgenden Arterien durchführen. Auch muss eine bildgebende Diagnostik des Gehirns mit Computertomographie (CT) und bei unauffälligem CT eine Kernspintomographie des Gehirns bei Erstdiagnostik vorgenommen werden. Dabei lässt sich dann meist die Ursache für einen Anfall finden, insbesondere Durchblutungsstörungen oder auch Hirntumore oder Gefäßanomalien. Auch venöse Blutungen zwischen Schädel und Gehirn (Subduralhaematome), die beispielsweise nach einem Sturz bei älteren Patienten auftreten, lassen sich damit beweisen.
Handelt es sich um Ohnmachtsanfälle und nicht um epileptische Anfälle, so ist eine zusätzliche intensive internistische und kardiologische Diagnostik notwendig. Hierbei müssen insbesondere Herzfunktionsstörungen, beziehungsweise Herzrhythmusstörungen auch im Zusammenhang mit gegebenen Medikamenten ausgeschlossen werden. Bei Flüssigkeitsmangel, ausgelöst durch Hitze oder fieberhafte Infekte, kann es auch zu Kreislaufkollaps und Ohnmachtsanfällen kommen. Auch erniedrigte Blutdruckwerte durch blutdrucksenkende Medikamente können eine Rolle spielen. Zahlreiche Medikamente können ebenfalls kreislaufbedingte Ohnmachtsanfälle oder epileptische Anfälle auslösen. Hier ist die Kontrolle und die Verträglichkeit der Medikamente untereinander notwendig. Auch Schwankungen des Blutzuckerspiegels im Rahmen von Diabetesbehandlung können Hirnfunktionsstörungen und Epilepsie, aber auch Kreislaufstörungen hervorrufen.
Übersicht über die wichtigsten Ursachen von Synkopen
Kardial (output)
- Herzvitien, insbesondere Aorten-, Mitralklappenstenosen, Embolien
- Myokardinfarkt, perikardiale Tamponade
- Bradyarrhythmie: Sick-sinus, AV-Block II. und III. Grades (Typ MOBITZ), ventrikuläre ES etc. Synkopen ohne Prodrome; (Glossopharyngeusneuralgie, Schlucken)
Tachyarrhythmie: supraventrikulär, ventrikulär QT-Verlängerung: Jerwell-Länge-Nielsen, Romano-Ward-Syndrom
Venöser Rückfluss
- VALSALVA-Manöver, Husten, Miktion, Vorhof-Myxom
Gehirndurchblutung
- Hypoxie, Hypovolämie, Anämie
- Extrakranielle oder intrakranielle Gefäßstenosen
- Migraine accompagnée
- Hyperventilationskonstriktion
Neurologische Grunderkrankung
- Hirnstamm (Syringobulbie), Multisystematrophie
- ALS
- Polyneuropathie, Guillain-Barré-Syndrom
- Autonome Funktionsstörungen
Internistische Ursachen von epileptischen Anfällen
Neben den Ohnmachtsanfällen durch Kreislauffunktionsstörungen kommt auch eine Reihe von internistischen Erkrankungen in Frage, die besonders im Alter zu epileptischen Anfällen führen. Bei Diabetes mellitus können Hypoglykämien oder nichtketotische Hyperglykämien sowohl fokale als auch generalisierte Anfälle auslösen. Nierenerkrankungen bei Dialyse oder chronischem beziehungsweise akutem Nierenversagen führen zu urämischer Enzephalopathie. Auch bei Leberzirrhose mit hepatischer Enzephalopathie treten Anfälle auf. Autoimmunerkrankungen des Bindegewebes (Kollagenosen) wie Lupus erythematodes, Sjögren-Symptom können zu Anfällen führen, ebenfalls Entzündungen der hirnversorgenden Gefäße, auch die sogenannte Wegener-Granulomatose, Polyarthritis nodosa und die rheumatoide Arthritis. Eine thyreotoxische Krise bei Hyperthyreose bei der Schilddrüse kann mit Anfällen einhergehen ebenfalls wie eine chronische Hypothyreose oder die sogenannte Hashimoto-Enzephalitis. Diese Ursachen sind nicht die häufigsten Auslöser für epileptische Anfälle, aber sie müssen bedacht und auf Hinweise für diese Erkrankungen muss geachtet werden. Am häufigsten sind allerdings cerebro-vaskuläre Durchblutungsstörungen die Ursache von Epilepsien ab dem 60. Lebensjahr und auch Hirntumoren ab dem 50. Lebensjahr bei Erstmanifestation eines Anfalls.
Ursachen für provozierte, epileptische Anfälle (Gelegenheitsanfälle)
- Hypoglykämie
- Hyponatriämie
- Entzug (Alkohol, Barbiturate, Benzodiazepine, AE)
- O2-Mangel
Die Erregungsschwelle senkende Pharmaka (Theophyllin, Phenothiazin, trizyklische Antidepressiva, Antihistaminika, Pentazocin)
Medikamentöse Therapie im Alter
Bei der Behandlung müssen insbesondere bereits bestehende Erkrankungen der älteren Patienten und deren medikamentöse Einnahmen bedacht werden. Es finden sich im Alter verminderte Aufnahme von Medikamenten, eine verminderte Nieren- und Leberfunktion sowie eine verminderte Serum-Eiweiß-Bindung. Dies ist insbesondere für die Medikamentengabe zu bedenken. Aber auch die veränderten Körperkompartimente wie Körperfett, Muskelmasse und Wassergehalt des Körpers beeinflussen die Wirksamkeit der Medikamente. Besonders wichtig ist die veränderte, meist vermehrte Rezeptorenempfindlichkeit für verschiedene Überträgerstoffe im Gehirn im Alter.
Aufgrund dieser Fakten empfiehlt es sich, eine langsamere Eindosierung, eine niedrigere Dauerdosierung, eine häufigere Kontrolle der Nebenwirkungen auf das Gehirn zu beachten.
Ursachen der veränderten Pharmakodynamik der Antiepileptika im Alter
- Einschränkung der Nierenfunktion
- Einschränkung der Leberfunktion und damit auch
- Verminderung des Albumins
- Erhöhte Rezeptorenempfindlichkeit im ZNS
- Erhöhung des Verteilungsvolumen der AE (Abnahme der Muskelmasse, Zunahme des Körperfetts)
Links
Links zu Kliniken mit Epileptologie in Ulm und um Ulm herum
Schwerpunktpraxis Epileptologie
Die Deutsche Gesellschaft für Epileptologie hat 1998 eine Arbeitsgruppe beauftragt, eine Definition von Schwerpunktpraxen zu erarbeiten. Das Anerkennungsverfahren haben folgende Praxen durchlaufen und können sich nun „Schwerpunktpraxen Epileptologie“ nennen.
Schwerpunktpraxen in Ihrer Region können über das Informationszentrum Epilepsie abgerufen werden.
Das Ausprägungsbild der Erkrankung
Überwiegend zwischen 58. – 60. Lebensjahr treten Erstsymptome auf, selten vor dem 30. Lebensjahr. Meist besteht anfänglich ein rhythmisches Zittern nur einer Hand oder auch eines Fußes, vor allen Dingen in Ruhe. Nach einiger Zeit verlangsamen sich Bewegungsabläufe und die Patienten werden etwas steifer, beim Gehen werden die Arme nicht richtig mitgeschwungen. Es kann schwerfallen, eine Bewegung zu beginnen oder sie auch abzubremsen. Die Patienten können z. B. aus einer sitzenden Position sich schlecht erheben. Der Gang wird zunehmend kleinschrittig und schlurfend, und es treten auch Gleichgewichtsstörungen und Störungen der Koordination beim Gehen auf. Schnelle Drehungen können nur langsam durchgeführt werden. Die Schrift wird kleiner, und die Mimik ist weniger lebhaft und wirkt maskenartig. Auch die Sprache wird monoton und leiser. Die Muskeln verhärten sich und bei Bewegungen kommt es zu muskulärem Widerstand. Häufig entwickeln sich Nacken- und Schulterbeschwerden. Die Symptome schreiten im Verlauf von 10 Jahren deutlich fort. Etwa nach 5 – 8 Jahren kommt es auch zu plötzlichen Unbeweglichkeiten (freezing), und Schwankungen der Symptome im Tagesablauf nehmen zu.
Morbus Parkinson - Definition und mögliche Ursache (Update 2020)
Beim Morbus Parkinson handelt es sich um eine neurodegenerative Erkrankung, bei der
es zu einem Untergang von Nervenzellpopulationen in verschiedenen Gehirnbereichen
kommt.
Besonders betroffen sind dopaminerge Neuronen in der Substantia nigra pars
compacta (ein Teil des Mittelhirns im Hirnstamm).
Als Folge kommt es zu einem Mangel des Botenstoffs Dopamin, dadurch zu einer
Verminderung der aktivierenden Wirkung der Basalganglien auf die Großhirnrinde und
schlussendlich zu einer Bewegungsstörung. Diese ist vor allem gekennzeichnet durch eine
Kombination aus Akinese (Bewegungsarmut) und mindestens einem der folgenden
Kardinalsymptome: Rigor („Steifigkeit“), Ruhetremor („Zittern“) und posturaler
Instabilität (Haltungsinstabilität).
Zusätzlich kann es zu einer Vielzahl an weiteren Begleitsymptomen kommen, u.a.
sensorischen Symptomen (Missempfindungen, Schmerzen, Verminderung des
Geruchssinns), vegetativen Symptomen (Störung der Kreislaufregulation, der Blasen- und
Darmfunktion sowie sexuellen Funktionen), psychischen Symptomen (Depression,
Schlafstörung) und kognitiven Einschränkungen.
Die Ursache für den Untergang der dopaminergen Neuronen ist noch ungeklärt.
Wie auch der Morbus Alzheimer gilt der Morbus Parkinson als alpha-Synucleinopathie.
Bei diesen Erkrankungen kommt es zu einer Ansammlung des Eiweißes alpha-Synuclein
in Nervenzellen. Dem gesunden Protein kommt eine wichtige Rolle bei der Regulierung
der Dopaminausschüttung zu. Bei Parkinson und Alzheimer ist das Protein, insbesondere
die Aminosäurenkette, falsch gefaltet. Dadurch können auch weitere Proteine nicht korrekt
abgebildet werden und es kommt zu einer Ansammlung alpha-synuclein-enthaltender
Lewy-Körperchen in den Nervenzellen.
Beim Parkinson betrifft das vor allem die Neurone in der Substantia nigra, aber tatsächlich
sind Lewy-Körperchen auch in den Enden der Riechnerven (bulbi olfactorii) sowie in
Nervengeflechten der Magen- und Darmschleimhaut zu finden.
Dies führte zu der Vermutung, dass der Morbus Parkinson sich eventuell vom Darm in das
zentrale Nervensystem ausbreitet, sowie gleichzeitig über die bulbi olfactorii ins Gehirn
gelangt (Dual-Hit-Hypothese).
In Tierversuchen mit Mäusen konnte gezeigt werden, dass es wenige Monate nach einer
Injektion fehlgefalteter alpha-Synuclein-Proteine in das Muskelgewebe von Dünndarm und
Magenausgang zu alpha-Synuclein-Ablagerungen im Hirnstamm kam. Auch konnte ein
Rückgang dopaminerger Neurone in der Substantia nigra nachgewiesen werden und die
Mäuse zeigten für einen Morbus Parkinson typische Bewegungsstörungen (Sangjune Kim
et al, 2019).
Bereits vorher war in mehreren Studien gezeigt worden, dass eine Durchtrennung
bestimmter Äste des N. vagus (wichtigster Nerv des parasympathischen Nervensystems
zur Regulation der inneren Organe) einen schützenden Effekt bzgl. des Auftretens eines
Morbus Parkinson hat (Liu B, Fang F, Pedersen NL et al, 2017).
Dies konnte in dem oben beschriebenen Tierversuch bestätigt werden. Die Mäuse, bei
denen der N. vagus direkt nach der alpha-Synuclein-Injektion durchtrennt wurde, zeigten
keine Ablagerungen und keine Parkinsonsymptomatik.
Die Beobachtung beweist noch nicht, dass der Morbus Parkinson im Darm entstehen
muss, sicherlich jedoch bezieht er den Magen-Darm-Trakt mit ein. Höchstwahrscheinlich
entwickelt sich der Morbus Parkinson mehrere Jahrzehnte vor dem Auftreten klinischer
Symptome unerkannt im Körper. Ein Ausbreitungsprozess entlang der Nerven Richtung
Gehirn ist ebenso möglich wie aus dem Gehirn Richtung Körper (vgl. Walter J. Schulz-
Schaeffer, Uniklinik Saarland).
Ebenfalls noch unklar ist, wie es zu der Fehlfaltung des alpha-Synucleins kommt.
Vermutlich spielen genetische Faktoren eine Rolle, ebenso wie Neurotoxine (Nervengifte).
Ein erhöhtes Erkrankungsrisiko nach Exposition zu manchen Pestiziden (z.B. Paraquat)
oder Lösungsmitteln (z.B. Trichloräthylen) ist bekannt. Interessanter Weise führt
Tabakkonsum zu einer Reduktion des Risikos.
Copyright © 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Verlauf und Häufigkeit
Erstsymptome treten zwischen 50. – 60. Lebensjahr meistens auf. Eine obere Altersgrenze für das Auftreten des Morbus Parkinson gibt es nicht. Die Häufigkeit der Erkrankung in der Bevölkerung (Prävalenz) liegt bei 100 – 200 Erkrankungen pro 100.000 Einwohner. Die Zahl der erkrankten Personen nimmt ab dem 50. Lebensjahr zu. In der Altersgruppe von 80 – 90 Jahren wurde eine Prävalenz von mehr als 1 % gefunden.
Jährliche Neuerkrankungen (Inzidenz) bei 10 – 20 auf 100.000 Einwohner. Zwischen dem 60. – 70. Lebensjahr kommen am meisten jährliche Neuerkrankungen dazu. Hier liegt die Inzidenz bei etwa 250 auf 100.000.
Die Parkinsonkrankheit schreitet stetig langsam fort und hat vor Einführung der Parkinsonbehandlung dazu geführt, dass fast ein Drittel der Patienten bereits 5 Jahre nach Krankheitsbeginn verstorben war. Seit Einführung der medikamentösen Behandlung (L-Dopa-Behandlung) hat sich die Mortalität von Parkinson-Patienten der Mortalität der Normalbevölkerung angenähert. Der Tod tritt bei der Parkinsonkrankheit in den meisten Fällen durch Infektionen, Insuffizienz des Herzens oder der Atmung ein.
Zellgifte als Ursache
Die Ursachen für den Schwund und den Abbau (Degeneration) von Nervenzellen im schwarzen Kern des Hirnstamms ist nicht gesichert. Man nimmt an, dass die betroffenen Nervenzellen untergehen weil sich schädigende Stoffe durch giftige Prozesse bilden. Diese Stoffe sind sogenannte freie Radikale, die anderen Molekülen Elektronen entreißen und es dadurch zur Zerstörung von lebenswichtigen Zellinhalten kommt. Insgesamt ist die Ursache aber nicht gesichert und man spricht daher bei der Parkinsonkrankheit von einem idiopathischen Parkinsonsyndrom (ca. 80 – 90 % der Fälle von Parkinsonsyndrom).
Bekannte Ursachen der Parkinsonkrankheit
Ist die Ursache bekannt, handelt es sich um ein symptomatisches Parkinsonsyndrom. Es kann nach einer Hirnentzündung auftreten. Auch Gifte können Parkinsonsyndrome verursachen, wie beispielsweise Mangan. Nach Kohlenmonoxydvergiftung (Vergiftung mit Auspuffgasen von Autos) können Parkinsonsyndrome entstehen. Ein Nebenprodukt in der Gewinnung eines Heroinersatzstoffes (MPTP) löst Parkinsonsymptome aus. Verschiedene Medikamente können Parkinson auslösen, insbesondere Medikamente, die zur Behandlung von Psychosen benutzt werden. Parkinsonsyndrome können auch nach Hirnschädigungen oder durch bestimmte Schlaganfälle beginnen.
Außerdem treten Parkinsonsyndrome nicht nur bei einer Degeneration der Bahnen zwischen schwarzem Kern und Streifenkörper auf, sondern ähnliche Bilder finden sich auch bei Zugrundegehen (Degeneration) anderer Hirnsysteme (Multisystematrophie und Untergruppen).
Diagnose
Die Diagnose erfordert mindestens zwei der drei Hauptsymptome (Unbeweglichkeit, Zittern, Muskelsteifigkeit). Andere neurologischen Symptome sollten nicht vorhanden sein oder sich auf eine unabhängige neurologische Krankheit zurückführen lassen. Ein gutes Ansprechen der Symptome auf Dopamin verstärkende Substanzen (L-Dopa) stützten die Diagnose. Zusätzliche einzelne Symptome bei der idiopathischen Parkinsonkrankheit treten unterschiedlich stark auf. Sie umfassen vermehrten Speichelfluss und Talgsekretion, Schweißstörungen, Verstopfung, Blasenstörungen, Erektionsschwäche, Depression, Verlangsamung des Denkens und verminderte Fähigkeit, Strategien und Denkkonzepte zu wechseln.
Medikamentöse Therapie
Dopaminersatz
Die Medikamente versuchen, den Mangel an Botenstoff der geschädigten oder untergegangenen Nervenzellfortsätzen des schwarzen Kerns beim Kontakt mit den Zellfortsätzen des Streifenkörpers zu ersetzen. Durch Zufuhr von L-Dopa kann der notwendige und mangelhaft vorhandene Überträgerstoff Dopamin wieder genügend hergestellt werden und seine Überträgerfunktion im Streifenkern wieder wahrnehmen. Dadurch verschwinden die Symptome des Parkinson-Syndroms. Auch kann die Bindungsfähigkeit des Überträgerstoffes an die Fasern des Streifenkörpers durch Medikamente verbessert werden (Stimulation der Dopaminrezeptoren durch Dopaminagonisten). Eine dritte Medikamentengruppe verhindert den Abbau von Dopamin an der Überträgerstelle durch Hemmung eines Abbauferments (Enzyms). Es handelt sich hier um Aminoxidasen, die gehemmt werden (Monoaminooxidase-B-Hemmung, MAO-B-Hemmer).
Glutamathemmung
Es kommt auch zur Beseitigung der Parkinson-Syndrome durch Einsatz von Substanzen, die nicht direkt an der Übertragung im Dopaminsystem beteiligt sind. Der Dopaminmangel führt zu einer Gegenregulation mit Überaktivierung von Überträgersystemen, die beispielsweise mit Glutamat, Acetylcholin oder auch Gaba funktionieren. Die Überaktivierung der Glutamatübertragung kann durch Amantadine reduziert werden, was zu einer Verbesserung der Parkinsonsyndrome führt.
Anticholinerge Wirkung
Die Überaktivierung der cholinergen Verbindungen bzw. Überträgerstellen wird mit Anticholinergika beeinflusst. Anticholinergika bewirken insbesondere eine Verbesserung des Zitterns.
Neben den genannten drei hauptsächlichen Wirkorten von Parkinsonmedikamenten gibt es für eine Reihe von Medikamenten noch nicht gesicherte Wirkmechanismen, insbesondere auch auf mögliche toxische Einflüsse in der Entstehung des Parkinson- Syndroms.
Unerwünschte Wirkungen von Anti-Parkinson-Medikamenten
L-Dopa, Dopaminantagonisten und Anticholinergika können zu Übelkeit, Erbrechen und Verstopfung führen. Psychische Nebenwirkungen bestehen bei L-Dopa, Dopamin- Antagonisten, Seligilin und Amantadin relativ ähnlich. Die Substanzen können zu Halluzinationen, Wahn, Unruhe, Verwirrtheit führen. Anticholinergika können ebenfalls Verwirrtheit und kognitive Störungen hervorrufen. Anticholinergika und L-Dopa können Harnverhalt, Erhöhung des Augeninnendrucks, tachykarde Rhythmusstörungen, Blutdruckerniedrigungen auslösen. L-Dopa, Dopaminantagonisten und Seligilin bewirken nach längerer Einnahme Dyskinesien (Störungen des Bewegungsablaufs mit Bewegungsunruhe).
Therapiemöglichkeiten bei Morbus Parkinson (Update 2020)
Zuerst kommen verschiedene Medikamente in Frage, die oral (über den Mund)
eingenommen werden. In erster Linie ersetzen diese Medikamente den fehlenden
Botenstoff Dopamin oder imitieren seine Wirkung (siehe auch oben).
Durch den Dopaminmangel kommt es zu einer Überaktivierung in anderen Bereichen,
sodass ein Überschuss bestimmter Botenstoffe (Glutamat, Acetylcholin) besteht. Dieser
Überschuss kann gehemmt werden, was ebenfalls die Parkinsonsymptomatik verbessert.
In den ersten Jahren der Therapie gelingt meist eine sehr gute Einstellung der Patienten.
Mit zunehmendem Krankheitsverlauf kommt es jedoch zu Wirkschwankungen der
Medikamente und damit zu Phasen sowohl mit ausgeprägter Bewegungsarmut als auch
mit guter Beweglichkeit (on/off-Phasen).
Bei ausgeprägten Wirkschwankungen können andere Therapieverfahren helfen:
- Apomorphin-Therapie: Apomorphin ist ein Dopaminagonist (wirkt also ähnlich
wie Dopamin) und kann entweder mit einem Pen unter die Haut gespritzt oder
durch eine Pumpe kontinuierlich zugeführt werden. Durch die Pumpe können
Wirkschwankungen besser ausgeglichen werden. - L-Dopa-Pumpe (Duodopa): Auch L-Dopa kann kontinuierlich verabreicht
werden. Dazu wird es über eine Pumpe und einen Schlauch direkt in den
Dünndarm geführt und dort vom Körper aufgenommen. - Tiefe Hirnstimulation: Hierbei wird dem Pat. ein programmierbarer
Impulsgenerator („Hirnschrittmacher“) eingesetzt, der elektrische Impulse erzeugt
und über feine Stimulationselektroden in bestimmten Hirnarealen abgibt.
Grundsätzlich kommen diese invasiveren Therapieoptionen besonders bei Pat. in Frage,
die mit einer rein oralen Medikation trotz mehrfach über den Tag verteilter Einnahme unter
zu starken Wirkschwankungen leiden, insgesamt aber noch recht gut auf die Wirkstoffe
ansprechen und auch sonst körperlich wie geistig relativ fit sind.
Deswegen sollte rechtzeitig über die verschiedenen Therapieoptionen gesprochen
werden, nicht erst, wenn die Gesamtsituation nicht mehr zu ertragen ist und ggfs noch
zusätzliche Erkrankungen aufgetreten sind. Häufig wird der richtige Zeitpunkt leider
verpasst.
Weitere Information/Quelle:
Leitlinie DGN „Idiopathisches Parkinson-Syndrom“ 2016
Holger Grehl, Frank Reinhardt: Checkliste Neurologie, Georg Thieme Verlag, Stuttgart
2016
A. Hufschmidt, C.H. Lücking, S. Rauer: Neurologie compact, Georg Thieme Verlag,
Stuttgart 2013
Copyright © 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Literatur
- R. Thümler:
Die Parkinson-Krankheit: Antworten auf die 152 häufigsten Fragen.
– Hilfreiche Informationen für Betroffene und Angehörige – , Stuttgart TRIAS, Georg Thieme Verlag, 1998. ISBN 3-89373-437-6 - Schering Lexikon „Morbus Parkinson“, Hrsg. Gsell et al, Stuttgart. Aesopus Verlag, 1997.
ISBN 3-773-1705-5 - T. Klockgether und W.H. Oertel:
Parkinson-Syndrome. S. 847 – 880: Therapie und Verlauf neurologischer Erkrankungen – Brandt, Dichgans, Diener (Hrsg.).
3. überarbeitete und erweiterte Auflage, Kohlhammer Verlag, 1998.
ISBN 3-17-015144-4 - Bochumer Therapiekonferenz zum Morbus Parkinson 1998. In: Aktuelle Neurologie.
4 Supplement, Band 25, Dez. 1998, S. 251 – 346. Organ der Deutschen Gesellschaft für Neurologie. - J. Volkmann, V. Sturm, H.J. Freund: Die subkortikale Hochfrequenz-Stimulation zur Behandlung von Bewegungsstörungen. In: Aktuelle Neurologie 25, 1998,
S. 288 – 296, Georg Thieme Verlag. - Pharmakotherapie der Parkinson-Krankheit. Hrsg. W.H. Oertel, Schatthauer Verlag 1999.
ISBN 3-7945-2015-7 - B.H. Yudim, B. Riederer: Freie Radikale und die Parkinson-Krankheit. Spektrum der Wissenschaft. 3/97, S. 52 – 61
Definition (Update 2020)
Als Schlaganfall (Apoplex/Insult) bezeichnet man eine durch eine Störung der Blutversorgung bedingte, plötzlich auftretende Funktionsstörung des Gehirns.
Ursächlich für einen Schlaganfall können sowohl Durchblutungsstörungen als auch Blutungen in das Gehirn sein. Eine Differenzierung beider Zustände, die völlig unterschiedliche therapeutische Konsequenzen haben, ist ohne bildgebende Verfahren nicht möglich.
Ischämische Insulte (also eine Durchblutungsstörung) machen mit ca 80-85% aller Schlaganfälle bei weitem die Mehrheit aus, intrazerebrale Blutungen (Blutungen im Gehirngewebe) sind in 10-15% ursächlich, Subarachnoidalblutungen in 5%.
Inzidenz: 140-200/100 000 Einwohner/Jahr, Prävalenz 600/100 000. 30% der Patienten versterben innerhalb 1 Jahres.
Einteilung nach Zeitdauer und Entwicklung:
- TIA (transitorische ischämische Attacke): Komplette Rückbildung der Symptomatik nach längstens 24h (häufig innerhalb 1h) und es kann keine Läsion in der Bildgebung nachgewiesen werden.
- Ischämischer Schlaganfall: Keine oder unvollständige Rückbildung der Symptomatik innerhalb 24h und/oder Läsionsnachweis.
- Ältere Definitionen wie PRIND sind obsolet, auch die Klassifikation als TIA ist wegen der heutigen Therapieoptionen und immer besser werdender bildgebender Verfahren zunehmend umstritten.
Ätiologie:
- Makroangiopathie (krankhafte Veränderungen größerer Gefäße) der extra- und intrakraniellen Gefäße: 20-40%, meist arterioarterielle Embolien; arteriosklerotisch (ACI-, Vertebralis-, Subclavia- oder intrakranielle Stenosen) und nichtarteriosklerotisch (Dissektion, Vaskulitis, Moya-Moya)
- Zerebrale Mikroangiopathie (krankhafte Veränderung kleinster Gefäße): 20-40%, arteriosklerotisch; meist lakunäre Infarkte, subkortikale Encephalopathie
- Proximale Emboliequelle: 25-40%, am häufigsten vom Herz ausgehend bei Vorhofflimmern, Koronarer Herzkrankheit, Endokarditis, Herzklappenfehler oder persistierendem Foramen ovale (s.u.)
- Gerinnungsstörungen: < 5%, häufig junge Patienten, genetisch (AT-III-Mangel, Faktor-V-Mutation, etc) oder erworben (Sepsis, Polytrauma, paraneoplastisch, Intoxikation)
Copyright © 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Akutbehandlung (Update 2020)
Die intravenöse Thrombolyse (Auflösung des Blutgerinnsels) mit Alteplase (rt-Pa) wird
innerhalb eines 4,5-Stunden-Fensters ohne obere Altersgrenze zur Behandlung
ischämischer Hirninfarkte empfohlen. Der Vorteil der Lysetherapie ist zeitabhängig, nach
wie vor gilt: Time is brain!
Die intravenöse Thrombolyse mit rt-PA kann bei selektierten Patienten auch zwischen 4,5
und 6 Stunden nach Symptombeginn als individueller Heilversuch zur Anwendung
kommen.
Erweiterte Bildgebungsparameter (z.B. Mismatch-Bildgebung, Kollateraldarstellung)
sollten herangezogen werden, um Patienten mit Risikogewebe (Gehirngewebe, das
betroffen, aber noch nicht verloren ist) zu identifizieren.
Bei unbekanntem Symptombeginn (z.B. Bemerken der Symptome nach dem Nachtschlaf)
kann durch bestimmte bildgebende Verfahren eine zeitliche Einschätzung erfolgen und der
Patient bei einem „DWI-FLAIR-Mismatch“ als Zeichen für einen frischen (<4,5h alten)
Infarkt noch lysiert werden (WAKE-UP-Studie 2018).
Eine Thrombolyse ist nicht möglich/empfohlen bei:
- Intrazerebraler Blutung (deswegen initiale Bildgebung mindestens mit nativer
kranieller Computertomographie nötig) - Frühe Infarktdemarkation (Abzeichnen von untergegangenem Gehirngewebe) in
einem erheblichen Anteil des Gehirns, das von der A. cerebri media versorgt wird
(pragmatisch ab > 1/3 des Mediaterritoriums, in amerikanischer Leitline > 1/3 der
Hemisphäre (!)). Grund: Risiko für Sekundärblutungen. - Schwerste klinische Beeinträchtigung (NIHSS >25, bzw Einzelfallentscheidung,
gilt nicht bei Infarkten im Basilarisstromgebiet) - Chirurgische Eingriffe oder relevantes Trauma in den letzten 30 Tagen
- Gerinnungsstörung mit Blutungsneigung; eine Vorbehandlung mit
Thrombozytenaggregationshemmern (z.B. Aspirin, Clopidogrel) ist keine
Kontraindikation, bei Vorbehandlung mit einer Antikoagulation gilt: je nach letzter
Medikamenteneinnahme, der Möglichkeit eines Antidots (Gegenmittel) und bei Vit-
K-Antagonisten des INR (bis 1,7 vertretbares Blutungsrisiko, siehe Xian 2012,
Mazya 2013) - nicht beherrschbarer Bluthochdruck (ab > 185/110mmHg trotz mehrfachem
Senkungsversuch). - Schlaganfall vor < 3 Monaten, nur in Ausnahmefällen bei kleinem Vorinfarkt bzw
Latenz > 6 Wochen bei schwerem neuen Defizit off-label; TIA zählt nicht als
Hinderungsgrund
Eine systemische Thrombolyse kann mit einer lokalen Thrombusentfernung
(Thrombektomie) kombiniert werden. Dies betrifft besonders proximale Verschlüsse
großer hirnversorgender Gefäße, z.B. Carotis-T-Verschluss oder L-Verschluss. Auch eine
intraarterielle Lyse mit oder ohne Thrombusentfernung in Kombination mit einer systemischen Lyse ist möglich, i.d.R. bis 6h. Im Basilarisstromgebiet, insbesondere bei
progressive Stroke (im Verlauf Verschlechterung der Symptomatik) im Sinne einer Ultimaratio-
Therapie auch später.
Effekte der Lysetherapie: Reduktion der Letalität nach 1 Monat von 15% auf 12%, nach 3
Monaten von 20% auf 17%, signifikante Reduktion der Morbidität (ein Drittel der Pat.
nach 3 Monaten unabhängig). In anderen Zahlen bzgl. eines guten Outcomes (modified
Rankin Scale <1): unter Lyse innerhalb von 90min wird der Prozentsatz an Pat. mit gutem
Outcome von 29% auf 41% erhöht, innerhalb von 91-180min Erhöhung von 30% auf 43%
(siehe auch Emberson 2014).
Für weitere Information/Quelle:
Leitlinie DGN „Akuttherapie des ischämischen Schlaganfalls – Ergänzung 2015“
Copyright © 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Sekundärbehandlung (Update 2020)
Sofern keine Indikation für eine Antikoagulation („Gerinnungshemmung“) besteht, sollte
innerhalb von 48h nach dem Schlaganfall (oder TIA) eine Sekundärprophylaxe mit einem
Thrombozytenaggregationshemmer eingeleitet werden.
Erste Wahl ist dabei ASS, bei Vorliegen von KHK, Angina pectoris, pAVK, Diabetes oder
z.N. Myokardinfarkt ist Clopidogrel erste Wahl. Die Einnahme erfolgt grundsätzlich
lebenslang, sofern im Verlauf keine Umstellung auf eine Antikoagulation notwendig wird.
Bei erneuter TIA oder Infarkt unter ASS kann ein Umstellen auf Clopidogrel erwogen
werden, ein Nutzen ist bisher aber nicht belegt.
Eine orale Antikoagulation ist u.a. bei einer kardialen Emboliequelle notwendig
(Vorhofflimmern oder -flattern, Herzwandaneurysma, offenes Foramen ovale), ebenso
nach Gefäßdissektion (bei letzterem i.d.R. für 6 Monate). Hierzu kommen vor allem die
neuen oralen Antikoagulantien (v.a. Dabigatran) in Frage, diese sind den herkömmlichen
Vitamin-K-Antagonisten vorzuziehen.
Unabhängig von Ausgangswert, Alter oder Geschlecht senkt die Gabe von Statinen zur
Cholesterinsenkung das Rezidiv-Risiko. Ziel-LDL mindestens < 100mg/dl.
Eine blutdrucksenkende Medikation ist, sofern nicht bereits vorhanden, ab dem
Initialereignis sinnvoll. In der Akutphase ist das Ausmaß der Blutdrucksenkung umstritten,
langfristig sollte der Blutdruck mindestens unter 140/90, besser auf 120/80 gesenkt
werden.
Bei symptomatischer Carotisstenose (Engstelle der Halsgefäße), also TIA oder Infarkt
gleichseitig zur Stenose innerhalb der letzten 6 Monate (absolute Indikation bei > 70%
Stenose, relative Indikation bei 50-70%) besteht eine Indikation zu Versorgung, entweder
operative Gefäßdesobliteration (Thrombendarteriektomie/TEA) oder Stenting.
Bei 20-25% der gesunden Bevölkerung liegt ein offenes (persistierendes) Foramen
ovale (PFO) als Verbindung zwischen beiden Herzvorhöfen vor.
Bei Patienten zwischen 16 und 60 Jahren mit einem (nach neurologischer und
kardiologischer Abklärung) unklaren ischämischen Schlaganfall und offenem Foramen
ovale mit moderatem oder ausgeprägtem Rechts-Links-Shunt sollte ein interventioneller
PFO-Verschluss durchgeführt werden (RESPECT-, CLOSE- und REDUCE-Studie)
Sollte ein Verschluss abgelehnt werden oder nicht möglich sein, ist eine Prophylaxe mit
ASS oder Clopidogrel zu empfehlen, eine Antikoagulation ist nicht als überlegen zu sehen,
sofern keine sonstige Indikation dafür besteht.
Nach einem PFO-Verschluss wird eine duale Thrombozytenaggregationshemmung mit
Aspirin plus Clopidogrel für 1–3 Monate empfohlen, gefolgt von einer 12–24-monatigen
Monotherapie mit Aspirin oder Clopidogrel. Bei Patienten mit zusätzlicher Manifestation
einer Arteriosklerose wird eine dauerhafte Monotherapie empfohlen.
Disclaimer: Sämtliche Medikamente dürfen nur vom Arzt verordnet werden und unterliegen
ständigen Kontrollkriterien. Sie sind rezept- und apothekenpflichtig. Diese Substanzen
dürfen auf der Grundlage der hier gegebenen Informationen nicht vom Leser oder durch
weitergegebene Informationen gekauft oder eingenommen werden. Der Autor übernimmt
keinerlei Verantwortung für die Nennung der therapeutisch wirksamen Medikamente auf
der Homepage.
Für weitere Information/Quelle:
Leitlinie DGN „Sekundärprophylaxe ischämischer Schlaganfall und transitorische
ischämische Attacke“
Leitlinie DGN „Kryptogener Schlaganfall und offenes Foramen ovale“
Copyright © 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Überblick
Definition
Beim Schlaganfall liegt eine rasche Gefühlsstörung oder ein Ausfall von Gehirnfunktionen durch Gefäßursachen vor. Die Funktionsausfälle halten beim Schlaganfall mindestens 24 Stunden an. Der Funktionsausfall des Gehirns beruht meist auf einer zu geringen Durchblutung im betroffenen Gebiet. Es kommt dadurch zu einem Sauerstoffmangel, der zu einem Zusammenbruch der Funktionen der Hirnzellen innerhalb von Minuten führt und schließlich auch zum Zelluntergang, zum Hirninfarkt.
Meist verursachen Gefäßverschlüsse eine verminderte Durchblutung durch Thromben (Blutgerinnsel). Auch durch Arteriosklerose (Alterungsvorgänge der Wände von Gefäßen bzw. Schlagadern) werden Verschlüsse ausgelöst. Thromben aus den Herzkammern können in die Hirngefäße gespült (embolisiert) werden und so zu Durchblutungsstörungen und Schlaganfall führen.
80 % aller Schlaganfälle sind Hirninfarkte durch Durchblutungsmangel. Neben Thrombosen und Arteriosklerose gibt es zahlreiche andere Ursachen für Schlaganfall. 15 – 20 % der Schlaganfälle entstehen durch Blutungen bzw. durch Platzen oder Zerreißen von Gefäßwänden von Schlagadern (intrazerebrale Blutung, Subarachnoidalblutung).
Symptome
Bei akutem Auftreten von neurologischen, teils auch psychischen Symptomen, wie Lähmungen, Gefühls-, Sehstörungen, Schwindel, Sprachstörungen, Gangunsicherheiten, akuten Gedächtnisstörungen oder Verwirrtheit, muss an Schlaganfall als Ursache gedacht werden und umgehend gehandelt werden. Der Schlaganfall ist wie der Herzinfarkt oder die Lungenembolie wie ein Notfall aufzufassen. Die umgehende Einlieferung in eine Schlaganfallstation (stroke unit) ist notwendig. Bis zu 3 Std. nach Beginn eines Schlaganfalls kann eine mit Medikamenten durchgeführte Auflösung gefäßverstopfender Blutgerinnsel (Thrombolyse) durchgeführt werden, danach erhöht sich das Risiko für die Thrombolysebehandlung. Dann war die Durchblutungsstörung zu lange anhaltend, als dass sich stark geschädigte Hirnzellen wieder erholen können.
Risikofaktoren und Epidemiologie
Diabetes mellitus, Bluthochdruck und Nikotinkonsum sind die wichtigsten therapeutisch beeinflussenden Risikofaktoren für Schlaganfall bzw. für die Alterung der Arterien. Bestehen diese Risikofaktoren, so erhöht sich bereits nach dem 50. Lebensjahr das Schlaganfallrisiko etwa alle 10 Jahre um das 2- bis 3-fache. Vorbeugung gegen Schlaganfall bedeutet Vorbeugung gegen die Risikofaktoren durch gesundes Leben mit Anpassung in Lebensführung, Nahrungs- und Essensgewohnheiten und Erreichen seelischer Gesundheit. Bluthochdruck, Diabetes mellitus, Übergewicht sollten konsequent und erfolgreich medikamentös behandelt und abgesichert werden.
Die Abnahme des Schlaganfallrisikos durch gerinnungshemmende und cholesterinsenkende Medikamente sowie durch die gute medikamentöse Einstellung eines Bluthochdrucks oder eines Diabetes mellitus ist messbar. Nikotinabstinenz führt zur Abnahme des Schlaganfallrisikos bereits nach einigen Monaten.
Folgen
Die Auswirkung der durch die Mangeldurchblutung ausgelösten Hirnfunktionen ist häufig schicksalhaft. Ca. 10 % der Betroffenen sterben nach einem Hirninfarkt. Bei Hirnblutungen liegt die Letalität (Sterberate) zwischen 40 – 50 % der Fälle. Bei ca. 20 – 25 % bleiben schwerwiegende neurologische Defizite auch nach erfolgreicher Rehabilitationsbehandlung bestehen. 25 – 30 % heilen unter Therapiebedingungen aber so aus, dass mit den zurückgebliebenen leichtgradigen Behinderungen die Verrichtungen des alltäglichen Lebens ohne höhergradige Einschränkung gemeistert werden können. Bleibt allerdings Pflegebedürftigkeit, so führt Schlaganfall zu langfristigen Lebensveränderungen – Belastungsfolgen bei Ehepartnern und in den Familien. Angehörigen von Schlaganfallpatienten kommt eine sehr wichtige Bedeutung in der Rehabilitation zu. Psychosoziale Betreuung in der Schlaganfallnachsorge ist notwendig.
* die Aufarbeitung neuerer Ergebnisse und Leitlinien ist in Arbeit
Epidemiologie (Ausbreitung und Häufigkeit)
In Deutschland ereignen sich pro Jahr etwa 185.000 – 220.000 Schlaganfälle. Summiert man die Zahl der Patienten über die Jahre, dann müssen etwa 800.000 Patienten auf Grund der Folgen von Schlaganfällen versorgt werden. Der Schlaganfall ist die häufigste neurologische Erkrankung und steht an Platz 3 der Todesursachen in Deutschland nach Herzerkrankungen und Krebs. Die Inzidenz der Erkrankung liegt in Deutschland bei 136, in Großbritannien bei 124 und in Frankreich bei 101 erstmaligen Schlaganfällen bezogen auf 100.000 Einwohner (Zeitraum 1995 – 1997). In einer Alterskategorie 60 J. und älter liegt die Inzidenz bei 160 erstmaligen Schlaganfällen auf 100.000 Einw., in der Alterskategorie 50 – 59 J. liegt sie nur halb so hoch bei 80 erstmaligen Schlaganfällen auf 100.000 Einwohner. Die Inzidenz des Schlaganfalls bei den 84jährigen beträgt 2.117 erstmalige Schlaganfälle auf 100.000 Einwohner. Die Schlaganfallhäufigkeit steigt also mit zunehmendem Alter an. Es muss durch die Zunahme der Lebenserwartung mit einem deutlichen Anstieg der Schlaganfallhäufigkeit gerechnet werden. So soll im Jahr 2030 der Anteil der über 65jährigen ca. 33 % betragen. (Daten nach Koluminsky-Rabas und Heuschmann 2002.)
Hypertonie
Der wichtigste einzelne Risikofaktor des Schlaganfalls ist die Hypertonie. Das Risiko für Schlaganfall ist direkt abhängig von der Höhe des Blutdrucks. Insbesondere für Hirnblutungen besteht ein 4 – 5-faches Risiko bei Vorliegen einer Hypertonie. Auch im höheren Lebensalter ist die Häufigkeit von Schlaganfall durch Hypertoniebehandlung verringert worden. Eine leichte Hypertonie liegt ab einem Wert größer 140/90 mmHg vor. Etwa 40 % der Schlaganfälle werden durch einen erhöhten Blutdruck erklärt (Weih et al. 2004).
Rauchen
Generell steigert Zigarettenrauchen das Schlaganfallrisiko gegenüber Nichtrauchern um 50%. Starke Raucher erhöhen ihr Risiko nochmals um das 2fache (starke Raucher: über 40 Zig./Tag). Das Risiko bei Frauen ist höher als für Männer. Nach Aufgabe des Zigarettenrauchens sinkt das Risiko für Schlaganfall. Das Risiko für Schlaganfall bleibt aber bei Ex-Rauchern erhöht, Weih et al. 2004. Rauchen ist auch ein besonderes Risiko für Subarachnoidalblutungen im Bereich Schädelbasisarterien (circulus Willisi), das Risiko für Subarachnoidalblutungen ist verdreifacht.
Herzerkrankungen
Das höchste Risiko hat das Vorhofflimmern. Das Vorhofflimmern ist die häufigste Herzrhythmusstörung und bewirkt Embolien. Bei Embolien werden Blutklumpen (geronnenes Blut) mit dem Blutstrom in den Adern fortgespült. Schlaganfälle durch Embolien bei Vorhofflimmern sind meist schwer und führen zu erheblichen Behinderungen oder auch Tod (Weih et al. 2004.) Die Häufigkeit von Vorhofflimmern erhöht sich alle 10 Jahre. 35 % aller Menschen mit Vorhofflimmern erleiden während ihrer Lebenszeit einen Schlaganfall. Die Veränderung der Herzklappen (Verkalkung der Mitralklappe), koronare Herzkrankheit, Herzinsuffizienz und links-ventrikuläre Hypertrophie stellen zusätzliche Risiken dar. Insbesondere bei Patienten unter 50 J. und bei Frauen kommt es auch durch ein offenes Foramen ovale zum Schlaganfallrisiko
Diabetes mellitus
Erhöhter Blutzuckerspiegel und Verminderung der Insulinrezeptorenqualität bzw. der dadurch ausgelöste Hyperinsulinismus führen zur Arteriosklerose der Gefäßwände. Das Risiko für Schlaganfall ist bei Diabetikern um 2 – 3 erhöht. Dabei ist dieses Risiko auch unabhängig von den häufig begleitenden zusätzlichen Risikofaktoren Hypertonie, Adipositas und Dyslipoproteinämie.
Übergewicht (Adipositas)
Auch für Adipositas besteht ein unabhängiges Risiko. Meist ist Übergewicht aber in einer Symptomkombination aus Adipositas, Hypertonie, Dyslipidämie, Hyperinsulinämie mit einem erhöhten Risiko behaftet. Gesamtcholesterin als auch LDL-Cholesterin verstärken die Arteriosklerose der A. carotis. HDL-Cholesterin vermindert die Arteriosklerose. Bei Hochrisikopatienten können Medikamente, die die Cholesterinspiegel senken, die Schlaganfallhäufigkeit um 30 – 46 % vermindern. Cholesterinwerte größer als 310 mg/dl erhöhen das Schlaganfallrisiko
Alkoholkonsum
Mit starkem Alkoholkonsum steigt der Risikofaktor beträchtlich. Oberhalb von 50 g Alkohol pro Tag (mehr als 1l Bier oder ½ l Wein findet sich das 5 – 11-fache Risiko für Hirnblutung. Für den Hirninfarkt durch Gefäßverschluss sind die Ergebnisse nicht eindeutig. Sehr geringe Mengen Alkohol (1 Drink pro Woche) führen dagegen zur Risikoreduktion. Wechselnde Alkoholexzesse z.B. an Wochenenden sind Risikofaktor für Hirninfarkte, besonders für Blutungen (auch bei jüngeren Erwachsenen). Leichter Alkoholkonsum führt zur geringeren Arteriosklerose der Karotiden (Halsschlagadern) und der Schädelbasisgefäße (circulus Willisi). Dies gilt aber nicht für die kleinen intrazerebralen Arterien. Moderater Alkoholkonsum kann Lipoprotein A senken und HDL-Cholestrin steigern. Alkohol kann die Blutfließeigenschaften verstärken, es reduziert die Thrombozytenaggregation und verstärkt die Fibrinolyse. Es verbessert auch die Verformbarkeit der roten Blutkörperchen. Die aufgeführten schützenden Effekte von Niedrigdosis Alkoholkonsum müssen aber dagegen abgewogen werden, dass diese Mengen auch zu chronischem Alkoholkonsum und damit auch zum Alkoholismus bzw. zu Suchtfolgen führen. Ein mittlerer Anstieg des Alkoholkonsums um 1 Drink pro Woche führt zu 10% mehr Alkoholkranken.
Hormonsubstitution
Bei Frauen vor der Menopause, die Hormone als Kontrazeptivum (Verhütungsmittel) einnehmen, ist das Risiko erhöht. Dramatisch wird es, wenn die Frauen an einer Hypertonie leiden oder aktuelle Raucherin sind. Dann steigt das Risiko auf das 7 – 11-fache.
Die Hormontherapie nach der Postmenopause wird häufig eingesetzt, um klimakterische Beschwerden und Osteoporose zu vermindern. Hier führt die Östrogensubstitution zur Verminderung von LDL-Cholesterin und erhöht HDL-Cholesterin und hat damit einen schützenden Effekt nach älteren Studien. Neuere Ergebnisse zeigen einen Risikozuwachs.
Risiko bei Östrogeneinnahme
- Erhöhtes Risiko für Schlaganfälle bei Östrogengehalt >0.050 mg
- Kein erhöhtes Risiko bei Östrogengehalt <0.050 mg
- Risikozunahme bei Frauen über 35 J., gleichzeitigem Nikotinabusus, arterieller Hypertonie, Migräne, Gerinnungsstörungen mit erhöhter Gerinnbarkeit des Blutes
- Hormonersatztherapie (HRT) in der Postmenopause ist ebenfalls mit Schlaganfallrisiko verbunden:
- HRT erhöht das Schlaganfallrisiko nach der Menopause um 7/1000 pro Jahr.
- Ein stark erhöhtes Risiko bei gleichzeitiger Hypertonie und Nikotin zusätzlich.
Drogenmissbrauch
Kokain-Missbrauch ist ein bedeutender Faktor bei jungen Erwachsenen für Schlaganfall. Insbesondere Hirnblutungen sind bei jungen Patienten an die Einnahme von Kokain gebunden.
Soziale Faktoren
Mit steigender sozialer Schicht nimmt das Risiko für Herz-Kreislauferkrankungen (Rauchen, Übergewicht, hohe Cholesterin-Werte, Bluthochdruck) ab (Kunst et al. 1998).
Homocystein
Es besteht eine enge Beziehung zwischen hohen Homocysteinspiegeln im Blut und dem Ausmaß von Verengung der Karotisarterie bzw. der Gefäßwanddicke (Intima media Dicke). Homocystein kann zur Endothelschädigung führen. In wenigen Studien wird ein Zusammenhang zwischen Homocystein als Risikofaktor mit einer Dosiswirkungsbeziehung für Hirninfarkt nachgewiesen. Studien, die Reduktion von Schlaganfällen durch Behandlung der erhöhten Blutspiegel beweisen sollen, werden durchgeführt. Eine Zusammenfassung über Homocystein und neurologische Erkrankungen findet sich in dem Buch von C. Bohlander-Gouaille und T. Bottiglieri, 2003. Auf den Zusammenhang von erhöhten Homocystein und Alkoholkonsum sei hingewiesen (Bleich et al. 2003).
Multiples metabolisches Syndrom
Bei erhöhtem Blutdruck, Störung des Lipid-Stoffwechsels und des Glukose-Stoffwechsels, Adipositas addieren sich die Faktoren und wirken häufig gegenseitig verstärkend. Beispielsweise erhöht sich bei einem 70-jährigen Mann in einem 10-Jahres-Zeitraum das Risiko von 5,4 % auf 79 % für Schlaganfall, wenn dieser Patient bei bereits medikamentös gut behandelter Hypertonie zusätzlich eine koronare Herzkrankheit, ein Vorhofflimmern, eine Verdickung des linken Herzmuskels, ein Diabetes und Rauchen als Risikofaktoren vorliegen hat. Patienten mit Übergewicht haben ein in etwa verdoppeltes Schlaganfallrisiko (Weih et al. 2004).
Chronische Infektionen sind mit einem erhöhten Risiko von Arteriosklerose behaftet, also auch für Schlaganfälle.
Körperliche Inaktivität
Körperliche Aktivität hat einen schützenden Effekt vor Schlaganfall. Inaktivität stellt einen Risikofaktor dar (Lee et al. 1999, Weih et al. 2004).
Seltene internistische Ursachen
Entzündliche Gefäßerkrankungen, Gefäßwandablösungen, Stoffwechselerkrankungen, Gerinnungsstörungen mit verstärkter Gerinnbarkeit, rheumatische Erkrankungen, Kollagenkrankheiten, Herzwandentzündungen, Herzklappenersatz stellen eine zusätzliche Gruppe von Risiken für Schlaganfall dar.
Arteriosklerose
Wie sind Arterien aufgebaut?
Die Zellen des Körpers brauchen Sauerstoff. Ohne Sauerstoff würden sie bereits nach kurzer Zeit absterben. Sauerstoff wird über das Blut in den Arterien an die Körperzellen transportiert. Dabei fließt es durch dickwandige Arterien, die sich ähnlich wie ein Baum in Äste, Zweige stets feiner aufteilen und so sämtliche Körperzellen mit Blut bzw. Sauerstoff versorgen können. Die Wände dieser Gefäße bestehen aus verschiedenen Muskelschichten. So sind diese dickwandigen Muskelarterien schließlich aufgezweigt in kleine Ateriolen. Die Ateriolen verästeln sich in die sogen. Kapillaren (Gefäß mit dem Durchmesser eines Haars, Haargefäße).
Die Arterien sind Schläuche mit „Muskelwänden“. Die Innenhaut steht in Kontakt mit dem durchfließenden Blut. Sie ist eine Schicht aus abgeflachten Epithelzellen, die Tunica intima („Intima“). Dieser Zellschicht ist aufgelagert eine Außenschicht der Tunica intima aus Bindegewebe und Proteinfasern. Es folgt als mittlere Schicht die Tunica media, die aus mehreren übereinander gelagerten Muskelzellen und elastischen Fasern besteht. Sie ist bei den großen Arterien die dickste Schicht. Die Muskelfasern haben die Aufgabe, dem Druck (Blutdruck) im Gefäß standzuhalten und ihn zu regulieren. Dabei können die Gefäße sich erweitern oder auch durch die Muskelfasern verengt werden. Als Außenschicht eines Gefäßes folgt dann die Tunica elastica, die dem Gefäß Stützfunktion gibt. Diese Schicht führt auch die kleinen Gefäße, die die Muskelzellen „ernähren“ müssen, die Vasa vasorum.
Wie kommt es zur Arteriosklerose?
Bei der Arteriosklerose kommt es zu einer Veränderung der Gefäßwände. Beispielsweise reagiert bei erhöhtem Blutdruck die Tunica media, die die dickste Muskelschicht im Gefäß darstellt, mit Sättigung bzw. einem kräftigeren Gefäßmuskel um dem erhöhten Blutdruck standzuhalten. Dabei kann diese Muskelwand dann so dick werden, dass sie selber nur schlecht mit Sauerstoff versorgt wird und es nach Zelluntergang zu Narbenbildungen in dieser Muskelschicht kommt. Auch kann das Lumen der Arterie (innerer Hohlraum durch den das Blut strömt) durch die Verdickung der Muskelschicht so stark eingeengt werden, dass zu wenig oder gar kein Blut mehr durch dieses Gefäß fließen kann. Es können sich auch durch zu langsamen Fluss Blutklumpen (Koagel und Thromben) bilden, die das Gefäß verstopfen und den Blutfluss durch das Gefäß verhindern. Dies trifft bei Bluthochdruck besonders für die kleinen Äste der Arterien, die Ateriolen und Kapillaren, zu.
Werden im Blut zu große Mengen an Fett, Cholesterin und ähnliche Substanzen transportiert, dann dringen diese Fettsubstanzen in die Tunica intima der Gefäße ein und führen hier zu Stoffwechselveränderungen. Es kommt zum narbigen Umbau der Gefäßwände und zu Auflagerungen und Verdickungen der Gefäßinnenwand. Es bilden sich Fettkerne und andere Ablagerungen (Atherome oder Plaques). Auch sind die Intaktheit und die Funktionsfähigkeit der Gefäßinnenwand von anderen im Blut transportierten Funktionen abhängig. Beispielsweise führen Substanzgruppen oder Moleküle (freie Radikale) zu toxischen Prozessen an den Zellen der Gefäßinnenwand und damit zu einem vernarbenden arteriosklerotischen Ablauf an der Innenwand der Gefäße mit Verdickung. Bakterien können in die Gefäßauskleidung bzw. in die verdickten Wände mit Atheromen und Plaques eindringen bzw. sich einnisten und die Arteriosklerose beschleunigen. Insgesamt sind die Gefäßinnenwand und auch die Muskelschicht in der Tunica media verschiedensten krankhaft wirkenden Prozessen unterworfen, die zur Versteifung der Gefäßwand, zur Verdickung und zu Auflagerungen und Verengungen führen. Mit zunehmendem Alter ist dies ein natürlicher Prozess, der durch die verminderte altersbedingte Regenerationsfähigkeit der beteiligten Zellen hervorgerufen wird.
Vorgänge bei Arteriosklerose
- Sättigung der Tunica media (Muskelschicht) durch erhöhte Druckanforderung (Hypertonie)
- Narbiger und bindegewebiger Umbau der verdickten Muskelschicht, teils mit Sauerstoffmangel in den verdickten Muskelzellschichten der Media
- Fett- und Cholesterin-Auskleidung der Gefäßinnenwand und Eindringen dieser Substrate in die Gefäßwand
- Giftige (toxische) Einflüsse auf die Zellen der Gefäßinnenwand mit Zelluntergang und narbigem Umbau
Was sind die Folgen der Arteriosklerose?
Die Fettablagerungen oder Plaques haben einen unterschiedlich rauhen bzw. narbigen Aufbau. Das Blut strömt an diesen Verhärtungen, Krusten und rauhen Oberflächen vorbei, und es kommt zur Ansammlung von Blutblättchen (Thrombozyten, die die Blutgerinnung in Gang setzen) und auch Blutzellen. Auf diese Weise bilden sich an Plaques und Atheromen Blutgerinnsel. Diese verengen das Lumen des Gefäßes zusätzlich und es kann zu einem zu geringen Durchfluss durch das Gefäß kommen. Gefährlicher ist es, wenn diese Thromben das Gefäß vollständig verstopfen. Es kann zum Ablösen des Blutklumpens oder Blutgerinnsels (Embolisierung) kommen, das dann mit dem Blutstrom in andere Gefäße gespült wird und diese verstopft. Die Blutgerinnsel können sich teilweise wieder auflösen, meist werden sie aber verfestigt und schließlich in die Gefäßwand „eingebaut“, so dass es zu zusätzlichen Vernarbungen und Unregelmäßigkeiten der Innenwand der Gefäße mit Verengungen kommt. So führt eine bereits begonnene Arteriosklerose zusätzlich zu einem verstärkten Fortschreiten der Arteriosklerose selber.
Im Kapillarnetz führen Wandverdickungen zu einem Untergang von den kleinen Kapillargefäßen und damit zu einer Verarmung in der Verästelung des Kapillarnetzes. Dies bewirkt Sauerstoffschädigungen an einigen oder auch größeren Zellgruppen und Absterben von Hirnzellen.
Die Sklerose der Arterienwände führt auch zu einer verminderten Elastizität und es kann zum Reißen der Gefäßwände kommen.
Daraus ergeben sich die klinischen Folgen der Arteriosklerose
- Kurzzeitige Verstopfung eines Gefäßes mit Auflösung eines Blutgerinnsels (vorübergehende ischämische Attacke mit kurzzeitigem zurückgebildetem Funktionsverlust)
- Verstopfung (Verschluss) einer Arterie mit Funktionsverlust und Zelluntergang der zu versorgenden Gewebe, irreversibel (ischämischer Schlaganfall)
- Schlechtere und gröbere Verästelung des Kapillar- und Ateriolennetzes mit langsamer, stetig abnehmender Blutversorgung der Gewebe (langsame Funktionsverluste)
- Bruch der Gefäßwand mit arterieller Hirnblutung. Überwiegender Untergang des vom ausgetretenen Blut abgedrückten und umspülten Gewebes (Hirnblutung)
Wo sind bevorzugte Orte (Lokalisation) von Arteriosklerose?
Arteriosklerotische Schädigungen der Gefäße kommen besonders an Aufzweigestellen und Gefäßabgängen vor, da hier der Blutfluss eine stärkere Belastung auf die Gefäßwände ausübt. Es wird davon ausgegangen, dass ein Drittel der arteriosklerotisch bedingten Gefäßverschlüsse an den Abgängen des Aortenbogens, ein weiteres Drittel an der Aufteilungsstelle der Halsschlagader (Karotisbifurkation) und ein letztes Drittel in den Gefäßaufzweigungen innerhalb des Kopfes ausgelöst wird.
Die durch die Gefäßbrüche ausgelösten Hirnblutungen finden meist in tiefen Hirnstrukturen (Stammganglien) statt.
Die Verarmung des Kapillarnetzes und das Zugrundegehen der Ateriolen führen zu Zellverlusten in Bereichen unterhalb der Hirnrinde (subkortikale arteriosklerotische Enzephalopathie) oder auch in den weißen Hirnregionen, die nahe zu den Ventrikeln (Hirnkammern) liegen.
Wie kann Arteriosklerose erkannt werden?
Mit sonographischer Methodik (Ultraschall) lässt sich das Aussehen der Gefäßwände und der Plaqueauflagerungen bildlich darstellen (Widder 1999). Die Dopplersonographie kann die Flussgeschwindigkeit in den Arterien messen. Aus der Flussgeschwindigkeit lässt sich gut auf den Verengungsgrad der Arterien schließen. Auch die Schichtung der normalen Gefäßwand kann sonographisch erkannt werden und Verdickungen, kleine Plaqueauflagerungen sowie langstreckige breite und dicke Gefäßauflagerungen können sonographisch dargestellt und in ihrer Größe vermessen werden. Der gefäßeinengende Effekt kann beurteilt werden. Am besten zugänglich für diese Ultraschalluntersuchungen (Doppler- und Duplex-Sonographie) sind die Halsschlagadern, also die A. carotis communis, A. carotis interna und A. carotis externa sowie die Nackenschlagader, die A. vertebralis. Es können Ultraschalluntersuchungen durch den Schädelknochen hindurch geführt werden und dabei Flussgeschwindigkeit und Flussrichtung von Arterien der Hirnbasis gemessen werden (transkranielles Doppler und transkranielles Duplex); Widder 1999.
Arteriosklerotische Verengungen der Gefäße können durch Röntgenuntersuchungen mit Kontrastmitteln, das in die Gefäße eingegeben wird, dargestellt werden oder auch über die Kernspinangiographie.
Wie wird Arteriosklerose therapiert?
Die Behandlung der Risikofaktoren einer Arteriosklerose ist das wichtigste Therapieprinzip. Arterielle Hypertonie, Fettstoffwechselstörungen, Diabetes mellitus, Tabakabhängigkeit sind die wichtigsten Risikofaktoren. Erhöhtes Homocystein (HyperHomocysteinämie) ist als Risikofaktor ebenfalls anerkannt.
Was ist die Aufgabe des Facharztes für Neurologie in diesen Fällen?
Die Ursache der aufgefallenen Symptome muss abgeklärt werden, insbesondere in der Hinsicht auf Arteriosklerose der hirnzuführenden Gefäße und der Gefäße im Gehirn selber. Das Risiko für einen Schlaganfall oder für eine Wiederholung der Symptomatik muss eingegrenzt und kompensiert werden. Die Risiken müssen entsprechend behandelt werden und der Patient sollte über das Maß seiner Arteriosklerose und das Ausmaß seines Risikos informiert werden. Da die Risiken insbesondere bei Hypertonie, Diabetes mellitus und Übergewicht, Bewegungsmangel wesentlich durch Lebensgewohnheit (Essgewohnheiten, Gesundheitsbewusstsein, Gesundheitspflege usw.) beeinflusst werden, ist auch die weitere Betreuung der Patienten zur Veränderung der Lebensgewohnheiten zu empfehlen.
Der Neurologe wird in Zusammenarbeit mit dem Hausarzt und möglicherweise einem Internisten oder Kardiologen die Ursachen der Durchblutungsstörungen abklären und beschreiben. Dafür ist die Angabe der Internistischen Risikokrankheiten wie Hypertonie, Hyperlipidämie, Übergewicht, Herzkrankheiten notwendig. Zusätzlich wird der Neurologe mit Ultraschallsonographie (Duplex und Doppler) den Grad der Arteriosklerose der hirnzuführenden Gefäße und auch Verengungen oder Verstopfungen von diesen Gefäßen finden und beschreiben können. Computertomographie und Kernspintomographie des Gehirns sichern die Diagnose ab. Insbesondere bei der Veränderung der kleinen Gefäße (Mikroangiopathie), die zu langsam zunehmenden neurologischen Defiziten bei älteren und bei Risikopatienten führt, werden sich deutliche Auffälligkeiten im Computer- oder Kernspintomogramm des Gehirns finden. Auch wird der Neurologe nach seltenen Ursachen von Schlaganfall bei jüngeren Patienten unter 50 Jahren fahnden und hier sein spezielles Wissen zur Anwendung bringen.
Klinik des akuten Schlaganfalls
(ischämischer Insult)
Der Schlaganfall geschieht plötzlich. Nur manchmal kann eine begleitende und spezifische Vorläufersymptomatik festgestellt werden wie Abgeschlagenheit, fieberhafter Infekt mit Husten und migräneartige Kopfschmerzen. Vom akuten Schlaganfall spricht man dann, wenn es zu neurologischen Defiziten wie Lähmungen, Gefühlsstörungen, Schwindel, Sehstörungen, Sprachstörungen durch Untergang von Hirnzellen kommt, die nicht mehr im weiteren Verlauf zurückgebildet werden. Bilden sich die Symptome innerhalb von 24 Std. oder auch später zurück, handelt es sich um eine flüchtige Durchblutungsstörung (transitorisch-ischämische Attacke, TIA). Beim Schlaganfall mit guter Rückbildung mit nur noch minimalen und nicht behindernden Symptomen ohne Zeitlimit handelt es sich um PRIND (partiell reversibles ischämisches neurologisches Defizit). TIA und PRIND können einem akuten vollendeten Schlaganfall vorhergehen.
Grundlage des Funktionsverlustes der Hirnzellen ist ein verminderter Blutdurchfluss bzw. eine verminderte Sauerstoffversorgung durch verschiedene Faktoren. An erster Stelle stehen die arteriellen Embolien. Hier kommt es durch verschieden mögliche Verletzungen der Gefäßwand, insbesondere durch Plaques zu Thrombosen, die dann in die Verzweigung des jeweiligen Gefäßes hineingetragen werden (Embolisation) und Gefäße verstopfen. Solche Embolien können auch aus dem Herzen stammen (kardiale Embolien). Auch Verschlüsse und hochgradige Verengungen von kleinen Arterien führen zu kleinen Infarkten (Untergang von Hirnzellen durch Sauerstoffmangel) und langsame Abnahme der Hirnfunktionen. Sind verschiedene Gefäße, die das Gehirn versorgen, verengt, kann es bei Blutdruckabfall oder zu wenig Blutvolumen zu einem Schlaganfall kommen. Hier liegt das Schlaganfallgebiet meist in der Grenzzone zwischen zwei versorgenden Hirnarterien (Grenzzoneninfarkt).
Die häufigsten Ursachen beim akuten Schlaganfall sind kardiale Embolien (25 %) gefolgt von Makroangiopathien (20 %) und kleinen Lakunärinfarkten (20 %). Der Rest setzt sich zusammen aus Schlaganfällen durch Gerinnungsstörungen, Gewalteinwirkung, Trauma, Entzündungen der Gefäßwände oder Fehleigenschaften des Blutes (hämatologische Erkrankungen).
Ursachen zerebraler Ischämien
| Ursache | Häufigkeit in % |
|---|---|
| Embolien aus Herz und Aortenbogen | 30 – 40 |
| arteriosklerotische Makroangiopathie | 20 – 30 |
| zerebrale Mikroangiopathie | 20 – 30 |
| seltene Ursachen (Gefäßwandablösungen, Gerinnungsstörungen, Gefäßentzündungen, u.a.) | 20 – 30 |
Verschiedene Orte des Schlaganfalls (Lokalisation)
Beim Schlaganfall gehen Zellen von Hirnregionen zu Grunde, die nicht mehr durchblutet werden. Da Funktionen des Gehirns unterschiedlich lokalisiert sind, wird der Verschluss einer bestimmten Hirnarterie zum Untergang von bestimmtem Hirngewebe und damit auch zu einem bestimmten Funktionsverschluss führen.
Tritt ein Schlaganfall beispielsweise im Stromgebiet der A. cerebri media auf, so kommt es zu gegenüberliegenden Halbseitenlähmungen mit zahlreichen anderen Symptomen. Ein Infarkt im Occipitallappen im Bereich des Hinterhaupts führt eher zu Sehstörungen, und beispielsweise ein Schlaganfall im Bereich des Kleinhirns zu Koordinationsstörungen.
Häufig lässt sich aus der Symptomkonstellation auf das Infarktareal und damit auch auf das verschlossene Gefäß zurückschließen. Das Schädigungsausmaß hängt zum einen von der Größe des Schlaganfalls ab, wobei sehr große Schlaganfälle zum Tode führen. Es können auch Hirnregionen von einem Schlaganfall betroffen werden, die sich im Alltagsleben nicht bemerkbar machen. Bei den so genannten strategischen Schlaganfällen handelt es sich um Schlaganfälle, die ein relativ kleines aber wichtiges Gebiet des Gehirns treffen und dadurch zu gravierenden neurologischen Defiziten führen.
Die Gesamtsterblichkeit bei Schlaganfall wird heute auf etwa 15% geschätzt. 40% der Patienten können nach dem Schlaganfall das Leben wie früher weiterführen. Patienten mit lakunären Infarkten (kleine Infarkte bei Mikroangiopathie) haben die beste Aussicht (Sterberate 3,3%).
Akutbehandlung
Die akute Behandlung des Schlaganfalls findet am besten in Krankenhäusern mit einer so genannten „stroke-unit“ statt. In solchen Abteilungen wird der Patient nach telefonischer Ankündigung oder als Notfall sofort kompetent diagnostiziert und behandelt. Diese „stroke-units“ sind bundesweit eingerichtet worden, damit die notwendige sofortige und schnelle Behandlung des Schlaganfalls tatsächlich durchgeführt werden kann. Dies hat bereits zu einer Verbesserung in der Behandlung und vor allem in den Schädigungsfolgen bei Schlaganfall geführt (Weimar und Diener 2003).
Nach Einlieferung eines Patienten in eine stroke-unit wird umgehend gemeinsam von Neurologen, Radiologen und Internisten die Diagnose gestellt und behandelt. Ziel der Diagnostik ist von neurologischer Seite zunächst festzustellen, ob es sich tatsächlich um ein neurologisches Defizit durch Schlaganfall handelt, in welchem Hirngebiet der Schlaganfall liegt, wie groß das vom Schlaganfall betroffene Hirngebiet ist und ob wichtige Lebensfunktionen beeinträchtigt beziehungsweise gefährdet sind. Dies gelingt durch die klinische Untersuchung und zusätzlich durch bildgebende Verfahren wie Computertomographie und Kernspintomographie. Auch lässt sich mit der Kernspintomographie und mit Ultraschall ein Gefäßverschluss oder eine verursachende Gefäßverengung häufig nachweisen.
Da ca. 30 % der Schlaganfälle durch Rhythmusstörungen des Herzens oder andere kardiologische Bedingungen verursacht sind, ist bei Hinweisen für eine solche Verursachung unverzüglich ein Kardiologe hinzuzuziehen.
Die Duplex- und Dopplersonographie der hirnzuführenden Gefäße und auch die transkranielle Duplexsonographie klären auf über die Blutversorgung in dem betroffenen Hirnareal. Auch kann man durch die transkranielle Doppler- und Duplexsonographie Hinweise dafür bekommen, wie mangelversorgte Hirnareale durch Umgehungskreisläufe oder von anderen Arterien neu beziehungsweise mitversorgt werden können.
Nach Einlieferung eines Patienten in eine „stroke-unit“ läuft parallel zur schnellstmöglich durchgeführten Diagnostik eine Primärversorgung des Patienten mit Basistherapie.
Basistherapie (pragmatisch)
- Systolische Blutdruckwerte nicht unter 160 mmHg senken
- Körpertemperatur nicht über 37,5° C
- Blutzucker zwischen 80 – 120 mg/% (bei Diabetikern 80 – 200 mg/%)
- Behandlung von Herzrhythmusstörungen
- Thromboseprophylaxe
- Bei schweren Schlaganfällen Intensivstation
- Lässt sich der Verschluss eines bestimmten Gefäßes im Gehirn diagnostisch sichern, wird unter bestimmten Bedingungen eine Auflösung des Embolus oder des Thrombus in dem verstopften Gefäß angestrebt (Thrombolyse).
Thrombolyse beim akuten Schlaganfall
- Gewebeplasminogenaktivator mit Wirksamkeit innerhalb der ersten 3 Stunden nach Schlaganfallbeginn (die Zeiten bei Prourokinase sind 6 Stunden bei selektiver Angiographie).
- Der beste Effekt 90 Min. nach Infarktbeginn.
- Nebenwirkung symptomatische zerebrale Blutungen.
- Lokale Thrombolyse.
- Eine Datenübersicht findet sich bei Külkens et al. 2004a.
- In speziellen Zentren kann auch mit Hilfe eines Katheters, der von einem Arzt über Röntgenbildgebung kontrolliert in das verstopfte Gefäß gesteuert wird, eine selektive Thrombolyse eines bestimmten Gefäßes durchgeführt werden (lokale Thrombolyse mit selektiver Angiographie), wie z. B. bei der A. cerebri media oder der A. basilaris.
- Die Wirkung der Thrombolyse (ohne selektive Angiographie) ist in verschiedenen Studien gesichert. Die Wirkung der Thrombolyse ist belegt für die ersten 3 Stunden nach Schlaganfallbeginn. Danach nimmt die Wirkung ab und ist statistisch in den verschiedenen Studien nicht mehr sicher. Aus diesem Grunde ist die wichtigste Bedingung zur Behandlung des akuten Schlaganfalls die möglichst schnelle Einweisung in eine stroke-unit. Dies sollte von den Angehörigen, von den Hausärzten und Neurologen bei einem Verdacht auf einen möglichen Schlaganfall umgehend durchgeführt werden.
- Findet sich keine Indikation für eine Thrombolyse, wird die frühe Sekundärprophylaxe notwendig; es muss ein Folgeinfarkt oder eine Vergrößerung des Infarktes verhindert werden.
Frühe Rehabilitationsbehandlung
- Krankengymnastik mit Mobilisierung
- Atemgymnastik, Behandlung von Schluckstörungen und Aphasie
- Training mit Hilfsmitteln
Frühe Depressionsbehandlung
- Positive, liebevolle Zuwendung
- Ermunterung zur Eigeninitiative
- Medikamente (Paroxetin, Citalopram, z. B.)
- Bleiben relevante neurologische Defizite, tritt durch den Schlaganfall eine Lebensveränderung des Patienten und seiner Umwelt ein. Hier ist weitere Therapie und chronische Behandlung notwendig.
Langfristige Lebensveränderungen, Belastungsfolgen
- Chronische rehabilitative Maßnahmen
- Gespräche und Psychotherapie, ggf. bei Angehörigen
- (psychosomatische Beschwerden der Angehörigen)
- Schmerztherapie
Fahrtauglichkeit bei Hirngefäßerkrankungen (Update 2020)
„Nach §2, Abs. 4 Straßenverkehrsgesetz (StVG), ist zum Führen von Kraftfahrzeugen
(nur) geeignet, wer die notwendigen körperlichen und geistigen Anforderungen erfüllt
und nicht erheblich oder nicht wiederholt gegen verkehrsrechtliche Vorschriften oder
gegen Strafgesetze verstoßen hat.“ (Positionspaper DGN Fahrtauglichkeit)
Wer nach § 315c des Strafgesetzbuches im Straßenverkehr ein Fahrzeug führt, „obwohl er
infolge geistiger oder körperlicher Mängel nicht in der Lage ist, das Fahrzeug sicher zu
führen […] und dadurch Leib oder Leben eines anderen Menschen oder fremde Sachen
von bedeutendem Wert gefährdet, wird mit Freiheitsstrafe bis zu fünf Jahren oder mit
Geldstrafe bestraft.“ (StGB)
Zusätzlich kann es versicherungs- und privatrechtliche Konsequenzen geben.
Jeder Autofahrer hat somit die Vorsorgepflicht und die Verantwortung, eigenständig zur
Sicherheit im Straßenverkehr beizutragen. Die Verantwortung kann nicht auf Dritte (z.B.
einen Arzt) abgegeben werden.
Eine Meldepflicht für neurologische, die Fahrtauglichkeit einschränkende Erkrankungen
besteht nicht.
Für Schlaganfall und TIA gilt zunächst grundsätzlich Fahruntauglichkeit, bis eine weitere
Abklärung und Risikoeinschätzung durchgeführt wurde, nach TIA mindestens für 6
Wochen, nach Schlaganfall mindestens für 3 Monate.
Es erfolgt eine Unterteilung in Gruppe 1 (PKW; Führerscheinklassen A, A1, A2, B, BE,
AM, L, T) und Gruppe 2 (LKW/Bus; Klassen C, C1, CE, C1E, D, D1, DE, D1E, FzF)
Laut Begutachtungsleitlinie für die Kraftfahreignung gilt: „Nach erfolgreicher Therapie
kann, abhängig von den besonderen Umständen des Einzelfalles, angenommen werden,
dass der Betreffende bedingt wieder in der Lage ist, Kraftfahrzeuge der Gruppe 1 zu
führen. Die Beurteilung setzt in der Regel eine stationäre Untersuchung voraus.“
Für die Gruppe 2 besteht nach Schlaganfall und TIA grundsätzlich keine
Fahrtauglichkeit, auch wenn kein persistierendes neurologisches Defizit vorliegt.
Über die Dauer der Fahruntauglichkeit für Gruppe 1 entscheidet ein qualifizierter Arzt,
i.d.R. ein Neurologe oder Verkehrsmediziner.
Die Fahrtauglichkeit kann ein Patient auf zwei Wege beurteilen lassen:
- Amtlicher Weg: Meldung bei der Fahrerlaubnisbehörde/Führerscheinstelle, diese wird eine Frist setzen, innerhalb derer ein verkehrsmedizinisches Gutachten, ein Nachweis über einen neuropsychologischen Test, ein Nachweis über eine Fahrprobe sowie ein medizinisch-psychologische Untersuchung eingefordert werden kann. Die Kosten für die Untersuchungen trägt der Patient selbst.
- Nicht-amtlicher Weg: Der Patient kümmert sich selbstständig um eine Belegung der eigenen Fahrtauglichkeit. Dafür sollte mindestens vorliegen: Ein Gutachten des behandelnden Arztes oder Verkehrsmediziners, ein neuropsychologisches Gutachten sowie ein augenärztliches Gutachten.
Der amtliche Weg ist notwendig bei Patienten, die zur Ausübung des Berufs auf ein Auto
angewiesen sind, sowie bei körperlicher Beeinträchtigung (ggfs Umbau des Autos
erforderlich). Grundsätzlich ist der amtliche Weg sicherer als der nicht-amtliche.
Wenn ein Patient ohne Überprüfung der eigenen Fahrtauglichkeit Auto fährt, erlischt der
Versicherungsschutz und es drohen strafrechtliche Konsequenzen. Es besteht eine
Vorsorgepflicht.
Für weitere Information/Quelle:
Positionspaper „Fahreignung bei Hirngefäßerkrankungen“, herausgegeben u.a. von DGN,
DGNB, DSG
Stiftung Deutsche Schlaganfallhilfe: „Autofahren nach Schlaganfall – Eine Anleitung zu
mehr Mobilität“
Copyright © 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Links
Information und Kontakt
- Praxis für Neurologie und Psychiatrie, Prof. Dr. med. K.P. Westphal, Neuer Graben 21, 89073 Ulm, Tel. 0731 66199
www.dr-k-westphal.de - Stiftung Deutsche Schlaganfallhilfe, Karl-Bertelsmann-Str. 256, Postfach 104, 33311 Gütersloh
www.schlaganfall-hilfe.de - andere Kontakte: www.kompetenznetz-schlaganfall.de
- Überwiegende Informationen dieses Artikels wurden entnommen:
Bücher: - Therapie und Verlauf neurologischer Erkrankung. 4. Aufl., Hrsg. T. Brandt, J. Dichgans, H.C. Diener, Kohlhammer Verlag, 2003
- Der Schlaganfall. Pathogenese, Klinik, Diagnostik und Therapie akuter zerebrovaskulärer Erkrankungen. Hrsg. A. Hartmann und W.D. Heiss, Steinkopfverlag Darmstadt, 2001
- Schlaganfall. Klinik, Diagnostik, Therapie. Hrsg. G.F. Hamann, M. Siebler, W. von Scheidt, Ecomed Verlagsgesellschaft, Landsberg, 2002
- Doppler- und Duplex-Sonographie der hirnversorgenden Arterien. 5. Auflage. Hrsg. B. Widder, Springer Verlag, 1999 (sechste Auflage 2004, im Druck)
- Leitlinien für Diagnostik und Therapie in der Neurologie. Hrsg. H.C. Diener bzw. Kommission „Leitlinien“ der Deutschen Gesellschaft für Neurologie, 2. Aufl., 2003, Georg Thieme Verlag, Stuttgart
- Frei von Tabak. Ein Stufenprogramm zur Raucherberatung und Rauchertherapie in der Arztpraxis. 3. Aufl., Hrsg. Bundesärztekammer in Zusammenarbeit mit der kassenärztlichen Bundesvereinigung. Bundesärztekammer Köln, 2001. ISDN 0945-1951
- Homocysteine: Related Vitamins and Neurophsychiatric Disorders. C. Bohlander-Gouaille und T. Bottiglieri, Berlin: Springer, 2003,ISBN 2-287-04393-4
- Übersichtsartikel und andere Zitate:
- Berger K. Epidemiologie cerebrovaskulärer Erkrankungen. In: Der Schlaganfall, Pathogenese, Klinik, Diagnostik und Therapie akuter cerebrovaskulärer Erkrankungen. Hrsg. A. Hartmann und W.-D. Heiss, Steinkopf Verlag Darmstadt, 2001, pp. 123-149
- Bleich S, Löffelholz K, Kornhuber J. Folsäure gegen Hyperhomocysteinämie. Ein neuer Ansatz zur Prävention und Therapie Alkoholismus-bedingter Störungen? Der Nervenarzt 2003; 5: 425-430
- Diener, H.C. und Hamann, G.F.: Primäre und sekundäre Prävention der cerebralen Ischämie. In: Therapie und Verlauf neurologischer Erkrankungen, 4. Aufl., Hrsg. Brandt, Th., Dichgans, J., Diener, H.C., 2003, pp. 359-476
- Heiss G, Sharret AR, Branes R, Chambless LE, Szklo M, Alzola C. Carotid atherosclerosis measured by B-mode ultrasound in populations: associations with cardiovascular risk factors in the ARIC study. Am J Epidemiol 1991; 134: 250-256
- Koluminsky-Rabas P u. Heuschmann PU: Epidemiologie des Schlaganfalls. In: Hamann, Siebler, von Scheidt, Schlaganfall. Klinik, Diagnostik und Therapie. Ecomed Verlagsges. Landsberg, 2002, pp. 25-45
- Külkens S, Ringleb PA, Hacke W. Empfehlungen der European Stroke Initiative (EUSI) zur Behandlung des ischämischen Schlaganfalls – Aktualisierung 2003. Teil 1: Organisation und Akutbehandlung. Der Nervenarzt. 2004a; 4: 368-379
- Külkens S, Ringleb PA, Hacke W. Empfehlungen der European Stroke Initiative (EUSI) zur Behandlung des ischämischen Schlaganfalls – Aktualisierung 2003. Teil 1: Prävention und Rehabilitation. Der Nervenarzt. 2004b; 4: 380-388
- Kunst AE, del-Rios M, Groenhof F, Mackenbach JP. Socioeconomic inequalities in stroke mortality among middle-aged men: an international overview. European Union Working Group on Socioeconomic Inequalities in Health. Stroke 1998; 29: 2285 – 2291
- Lee IM, Hennekens CH, Berger K, Buring JE, Manson JE. Exercise and risk of stroke in male physicians. Stroke 1999; 30: 1-6
- Sacks FM, Vetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER, Simons-Morton DG, Karanja N, Lin PH, for the DASH-Sodium collaborative research group. Effects on blood pressure of reduced dietary sodium and the dietary approaches to stop hypertension (DASH) diet. N Engl J Med 2001; 344: 3-10
- Schellinger PD, Jüttler E, Meyding-Lamadé Uta K, Schwark C. Stellenwert von Thrombozyten-Aggregations-hemmern in der Sekundärprävention des ischämischen Schlaganfalls. Fortschr Neurol Psychiat 2004; 72: 270-281
- Weih M, Müller-Nordhorn J, Amberger N, Masuhr F, Lürtzing F, Dreier JP, Hetzel A. Risikofaktoren des Schlaganfalls. Übersicht über die Evidenz in der Primärprävention. Der Nervenarzt 2004; 4: 324-335
- Weimar C und HC Diener. Diagnose und Therapie der Schlaganfallbehandlung in Deutschlang, Ergebnisse der Deutschen Schlaganfall Datenbank. Deutsches Ärzteblatt Jg. 100, Heft 40, 2003, pp. A2576-A2582
- Wirth A. Lebensstiländerung zur Prävention und Therapie von arteriosklerotischen Krankheiten. Deutsches Ärzteblatt 2004. Jg. 101; 24: A1745-1752
Migräne bezeichnet periodisch auftretende Attacken von Kopfschmerzen mit Begleitsymptomen. Bei einigen Patienten gehen der Attacke Vorläufersymptome voraus. Dies wird Aura genannt.
Typen
Man unterscheidet Migräne ohne Aura, Migräne mit Aura sowie komplizierte Migräne und kindliche Migräne.
Ursachen
Wahrscheinlich kommt es auf der Grundlage von entzündlichen Veränderungen an der Gefäßwand, die teilweise auch neurogen ausgelöst sein können, zur Entstehung von Migränekopfschmerz. Substanzen, die zu einer Verengung der Gefäße und gleichzeitiger Entzündungshemmung führen, sind deshalb besonders wirksam.
Tiermodell und Wirkmechanismen
Versuche an Tieren haben gezeigt, dass besonders die Nervenfasern des Trigeminusnervs und des Gesichtsnervs (Nervus trigeminus und Nervus facialis), die Arterien des Gerhirns und der Hirnhaut versorgen. Zusätzlich gibt es schmerzleitende Nervenfasern, die über die oberen Nervenwurzeln der Halswirbelsäule zu den Kernen des Trigeminusnervs im Hirnstamm geleitet werden. Durch Reizung von Fasern dieser Nerven kommt es an den Arterien und Venen zu Veränderungen, die einer Migräne entsprechen. So erweitern sich die Arterien der Hirnhaut und auch kleine Venen werden erweitert. Die Durchlässigkeit der Gefäße wird erhöht, Proteine aus Zellen, die im Blut vorhanden sind, treten aus. Überträgerstoffe von Schmerzfasern werden stimuliert. Eine neurogene Entzündung mit Gefäßerweiterung ist entstanden. Diese Phänomene werden durch Schmerzfasern an das Gehirn vermittelt und lösen im Bewusstsein das Erleben einer Migräne aus. Mit migränespezifischen Medikamenten (Sumatriptane) und auch mit Ergotaminen lassen sich die durch Reizung von Trigeminusfasern ausgelösten verstärkten Durchblutungen der Hirnhaut und des Gehirns hemmen. Auch das für die Schmerzmodulierung wichtige Calcitonin genbezogene Peptid (Calcitonin Gene Related Peptide CGRP) wird durch die Substanzen normalisiert. Diese Erkenntnisse aus Tiermodellen lassen ähnliche Wirkmechanismen für die menschliche Migräne annehmen.
Beschwerden
Migräneattacken und Migränekopfschmerzen laufen individuell unterschiedlich ab. Stärke und Art variieren. Migränen beginnen meist ohne Aura mit einer Art Druckgefühl über einer Kopfhälfte oder in der Augenregion. Häufig sitzt der Schmerz auch im Schulter- oder oberen Nackenbereich. Es kommt zur Verstärkung. Der Schmerz kann hämmernd, klopfend, bohrend und pulsierend sein oder auch dumpf zunehmend. Der Schmerz ist mit einem starken Unwohlsein verbunden. Konzentration und Aufmerksamkeit auf die Umgebung fallen schwer. Es entwickelt sich Übelkeit. Die Arbeitsfähigkeit ist vermindert oder fast nicht gegeben. Die Gesichtsfarbe wird blass oder rötlich, Hände und Füße sind schlecht durchblutet, Licht- und Lärmempfindlichkeit sowie Empfindlichkeit auf andere Reize und eine soziale Empfindlichkeit entstehen. Ruhe, Rückzug, Verdunklung, Lärmvermeidung werden gesucht. Körperliche Belastung, Husten, Niesen, Kopfbewegungen verstärken den Kopfschmerz in massiver Weise. Häufig kommt es zu Erbrechen und auch zu Geruchsüberempfindlichkeit.
Bei der Migräne mit Aura kommt es vor dem Schmerzbeginn zu Ausfallserscheinungen. Es treten Sehstörungen oder Sehen von Blitzen, Lichtern, Ringen, Kreisen oder Zacken auf. Auch können im Gesichtsfeld oft eine Hälfte oder Teile des Gesichtsfeldes nicht richtig wahrgenommen werden. Auch Lähmungen, Drehschwindel, Doppelbilder können als Aura oder Vorläufersymptome einer Kopfschmerzattacke auftreten. Die Vorläufersymptome (Aura) entwickeln sich innerhalb von Minuten und dauern meist nicht länger als 1 Stunde. Häufig kommt es zum überlappenden Übergang von Aurasymptomen in den Kopfschmerz. Die Aurasymptome werden mit Beginn des Kopfschmerzes deutlich weniger oder verschwinden.
Bei der komplizierten Migräne bleiben die Ausfallserscheinungen bestehen und man kann in seltenen Fällen in der Computertomographie oder Kernspintomographie kleinere Schädigungen der Hirnsubstanz durch Sauerstoffminderversorgung feststellen.
Diagnostik
Die Diagnose erfolgt im überwiegenden Maße durch die Schilderung des Anfallsablaufs, der Symptome und muss hier abgegrenzt werden gegen Spannungskopfschmerz, Cluster-Kopfschmerz, neuralgische Kopfschmerztypen oder auch arzneimittelinduzierten Kopfschmerz. Elektroenzephalographie (EEG) und Hirnpotentiale und ggf. Kernspintomographie zum Ausschluss gegenüber anderen Erkrankungen mit ähnlichen Ausfallserscheinungen sind notwendig. Die Untersuchungen erfolgen durch Neurologen.
Auslösefaktoren für Migräne
Verhalten und Umwelt können einen Migräneanfall auslösen, wie beispielsweise verqualmte Räume, Höhe, Kälte, Flackerlicht und Lärm. Schlafmangel, zu viel Schlaf bzw. Störungen des Schlaf-Wach-Rhythmus, Stress und Sorgen, körperliche Anstrengung, sportliche Aktivität mit Hochleistung, Hunger, Erwartungsängste, Entlastung nach Stress, Wochenendmigräne, Zeitverschiebungen. Hormonelle Faktoren spielen bei der Entstehung von Migräneattacken eine Rolle. Menstruation (Regelblutung), Eisprung, Verhütungsmittel („Pille“).
Zahlreiche Substanzen können Migräne auslösen, besonders Alkohol und Rotwein. Südfrüchte, Kakao (Schokolade) kakaohaltige Getränke und andere. Käsesorten, Glutamat, Nitrit, Salz, selten auch Fettspeisen.
Bluthochdruck (Hypertonie) als auch niedriger Blutdruck (Hypotonie) können zu erhöhter Migränehäufigkeit führen. Überlastungen der oberen Halswirbelsäule, Massage, Wärmeanwendungen, Medikamente wie Nitroglyzerin und Kalziumantagonisten lösen ebenfalls Migräneanfälle aus.
Seltene Faktoren zur Auslösung des Migräneanfalls sind: Luftfeuchtigkeit, Überdosis Vitamin A, kalte Speisen, Lesen, Gerüche, Parfum sowie Neonlichtbeleuchtung und allergische Reaktionen.
Seltene Beschwerden
Kurzatmigkeit, Schluckbeschwerden, Kloßgefühl, Schwäche, Völlegefühl, Abgeschlagenheit, Mattigkeit, Reizbarkeit, Grübelei, innere Unruhe, Schlafbedürfnis, Zittern, Nacken-Schultern-Schmerzen, Rückenschmerzen, Sehstörungen, Gleichgewichtstörungen, Schluckauf, Gähnen, Atemnot, Erstickungsgefühl, Neigung zum Weinen, Leibschmerzen, Angstgefühle, Bauchschmerzen, Harn- und Stuhlabgang, Glieder- und Gelenkschmerzen, Erröten, Frieren, kalte Füße, Hitzewallungen, Taubheitsgefühle, vermindertes sexuelles Bedürfnis oder Erregbarkeit.
Vorboten einer Migräne
Unabhängig von der Aura gibt es auch Vorboten einer Migräne mit Stimmungsschwankungen wie beispielsweise auffallende Heiterkeit oder Umtriebigkeit bis zu Trägheit, Teilnahmslosigkeit und Müdigkeit.
Behandlung der Migräne (Update 2020)
In der medikamentösen Behandlung gibt es für die akute Migräne sehr wirksame Substanzen; ebenso gegen chronische Migräne. Hier muss der Arzt konsultiert werden.
Grundlagen der medikamentösen Therapie (Update 2020)
Es wird zwischen einer Akutbehandlung bei Bedarf, also bei Auftreten einer
Migräneattacke und eine prophylaktischen Therapie unterschieden.
Für die Behandlung einer Migräneattacke kommen neben herkömmlichen, größtenteils
rezeptfrei erhältlichen Schmerzmedikamenten auch migränespezifische Medikament in
Frage. In erster Linie sind dies Triptane. Hiervon gibt es verschiedene Präparate, wie
auch verschiedene Applikationsformen (Schmelz- und Filmtabletten, Nasenspray, Pens
zur s.c. Gabe, etc). Welches Präparat am besten geeignet ist, entscheidet der
behandelnde Neurologe.
Eine reine Bedarfsmedikation ist dann ausreichend, wenn die Migräne nur selten auftritt
(maximal 2x im Monat) und bei Bedarf gut auf die Akutmedikation anspricht. Tritt die
Migräne häufiger auf (< 2 Tage/Monat) oder mehrere Tage am Stück, sollte eine
prophylaktische medikamentöse Behandlung erwogen werden. Auch bei fehlendem
Ansprechen der Attacke auf die Bedarfsmedikation oder zu starken Nebenwirkungen der
Akutmedikation ist eine Prophylaxe sinnvoll.
Werden an mehr als 10 Tagen/Monat Schmerz- oder Migränemedikamente eingenommen,
besteht die Gefahr eines medikamenteninduzierten Kopfschmerzes. Dieser macht sich
durch eine Zunahme der Kopfschmerzen bzw einer Zunahme der Migränehäufigkeit
bemerkbar. Es entsteht ein Teufelskreislauf aus zunehmenden Schmerzen und einem
(gefühlt) höheren Bedarf an Schmerzmitteln. Eine prophylaktische Therapie kann auch
hier Abhilfe schaffen und den Bedarf an Akutmedikamenten auf ein unproblematischeres
Maß senken.
Als Migräneprophylaxe kommen verschiedene Wirkstoffe in Frage, u.a. aus dem Bereich
der Antikonvulsiva, Beta-Blocker und Kalziumantagonisten. Seit kurzem sind auch
verschiedene Antikörper zur s.c. Applikation verfügbar. Verschiedene Kriterien fließen in
die Entscheidung zu einem Präparat ein, vor allem auch potentielle Nebenwirkungen.
Disclaimer: Sämtliche Medikamente dürfen nur vom Arzt verordnet werden und unterliegen
ständigen Kontrollkriterien. Sie sind rezept- und apothekenpflichtig. Diese Substanzen
dürfen auf der Grundlage der hier gegebenen Informationen nicht vom Leser oder durch
weitergegebene Informationen gekauft oder eingenommen werden. Der Autor übernimmt
keinerlei Verantwortung für die Nennung der therapeutisch wirksamen Medikamente auf
der Homepage.
Grundsätzlich gilt, dass eine Prophylaxe langfristig angelegt ist und eine Wirkung meist
erst nach mehreren Wochen oder Monaten erwartet werden kann. Die Medikamente
werden unter Rücksichtnahme auf Nebenwirkungen langsam eindosiert und bei guter
Verträglichkeit gesteigert. In der Regel kann man keine komplette Attackenfreiheit
erwarten, sondern bei gutem Ansprechen eine Reduktion der Attackenfrequenz um ca.
50%.
Die prophylaktische Therapie sollten mittels Migräne-Tagebuch überwacht werden. Falls
ca 2 Monate nach Erreichen der Enddosis keine Besserung eingetreten ist, empfiehlt sich
ein Therapiewechsel.
Eine medikamentöse Prophylaxe sollte immer in Kombination mit nicht-medikamentösen
Maßnahmen angewandt werden, z.B. regelmäßiger aerober Ausdauersport und
verhaltenstherapeutische Maßnahmen (Entspannungsübungen, Stressmanagement, etc).
Für weitere Information/Quelle:
Holger Grehl, Frank Reinhardt: Checkliste Neurologie, Georg Thieme Verlag, Stuttgart
2016
A. Hufschmidt, C.H. Lücking, S. Rauer: Neurologie compact, Georg Thieme Verlag,
Stuttgart 2013
Copyright © 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Nichtmedikamentöse Behandlung
Ruhe und Abschirmung insbesondere von Licht und Lärm. Hinlegen in abgedunkeltem Raum mit Kälteanwendung an Stirn und Nacken mit einem Lappen oder mit „Eisbrillen“.
Physikalische Methoden sind nur vorsichtig anzuwenden. Vorsichtige Lymphdrainage oder Massage des Gesichts und des betroffenen Kopfteils kann Linderung bringen, kann aber auch verstärken. Insbesondere Dehnung der Hals- und Schultergürtelmuskulatur kann verstärken, nur selten bessern.
Vorbeugende Migränebehandlung
Mit physikalischen Methoden wie Massage, Gymnastik, Jogging und Schwimmen. Auch Entspannungstraining nach Jacobsen (Relaxationstraining). Autogenes Training und Bio-Feedback-Methode helfen. Joga, Shiatsu und andere alternative Methoden haben bei der vorbeugenden Migränebehandlung häufig einen guten Effekt. Wichtig ist bei der nichtmedikamentösen vorbeugenden Migränebehandlung das Führen eines Migräne-Tagebuchs oder -Kalenders, um auslösende Faktoren zu erkennen und diese dann zu meiden.
Migränepatienten zeigen oftmals eine gesteigerte Empfindlichkeit gegenüber Stress. Konfliktsituationen im Beruf und Familie, Ehe, Sexualität sollten gelöst werden (psychotherapeutische Verfahren, Verhaltenstherapie).
Literatur*
- Migräne von H. C. Diener und V. Limmroth, S. 3-18. In: Therapie und Verlauf neurologischer Erkrankungen. 3. Auflage, Hrsg. Brandt, Dichgans, Diener.2003, Kohlhammer Verlag
ISBN 3-17-017926-8 - Migräne, eine Informationsbroschüre für Patienten der Firma Dolorgiet GmbH und Co KG, Otto v. Gehrike-Str. 1, 53574 St. Augustin-Bonn
- Schmerzsyndrome des Kopf- und Halsbereichs. Hrsg. Paulus und Schöps, Wissenschaftliche Verlagsges.mbH. Stuttgart, 1998, S. 19-50
- Pfaffenrath, Volker: Der chronische Kopfschmerz: Spannungskopfschmerz und Schmerzmittelmissbrauch. 3. Auflage, Arcis Verlag München, 1992
ISBN 3-89075-014-1 - Ensink, F.B.M; Sokya, D. (Hrsg.): Migräne: Aktuelle Aspekte eines altbekannten Leidens. Springer Verlag 1994. ISBN 3-540-55760-1
- Peikert, Andreas: Kopfschmerzen verstehen und erfolgreich behandeln.
Stuttgart TRIAS. Georg Thieme Verlag 1997. ISBN 3-89373-402-3*
* Neuere Therapie-Methoden zur prophylaktischen Therapie in Aufarbeitung
Aspekte der Chronifizierung
Das Halswirbelsäulen-Distorsions-Trauma stellt eine Beschleunigungsverletzung der Halswirbelsäule, ihrer Muskelbänder, Knochen, Nerven und Gefäße dar (Saternus, 1997). Der Schweregrad der Verletzung hängt von dem Ausmaß der Beschleunigung ab, wobei es sich meist um passive, scharf einsetzende Wirkungen auf den Kopfhalteapparat handelt. Es kommt zu negativen und positiven Beschleunigungen (Vor- und Rückschlag) wie beispielsweise bei einem Heckaufprall. Die HWS-Distorsionsverletzungen sind in ihrem objektiven Umfang, insbesondere bei leichten oder mittelgradigen Verletzungen, schwer zu erfassen; eine Primärverletzung insbesondere z. B. durch Mikrotraumata kann auch kernspintomographisch übersehen werden. Trotzdem bestehen auch ohne faßbare Primärverletzung typische Beschwerden und Verläufe. Ein besonderes Problem besteht bei chronischen Verläufen, bei denen es zu psychischen Fehlentwicklungen kommen kann (Kallert, 1995).
Symptome und Einstufung nach Schweregraden
An Symptomen treten nach M. Keidel (1995) Nackenschmerzen (100 %), Nackensteife (88 %), Kopfschmerz (87 %), vegetative Beschwerden (71 %), Halsmuskelschmerzen (70 %), neurasthenische Beschwerden (60 %), Kopfschwere (48 %), Schwindel (38 %), Armbeschwerden (27 %), Kreuzschmerzen (25 %), Hörstörungen (21 %), Handsymptome (20 %), Sehstörungen (20 %), Kloßgefühle im Hals (12 %), Schluckschmerz (7 %), rauher Hals (6 %), Kieferschmerz (4 %), Mundbodenschmerz (4 %) auf. Ähnliche Verteilungen finden andere Autoren (Gutmann, 1988).
Bei der neurologischen Untersuchung tritt insbesondere das Phänomen des „Muskelhartspanns“ auf, das bereits bei leichten Distorsionstraumata ausgelöst werden kann. Entsprechend findet sich ein ein- oder doppelseitiger Muskelhartspann mit Einschränkung der Kopfbewegungen sowie eine ausgeprägte Berührungsempfindlichkeit im Nacken. Dabei ist der Nackenschmerz nahezu bei jedem in der Akutphase ausgeprägt, 90 % der Patienten berichten über ein Gefühl von Nackensteife. Der Nackenschmerz strahlt in benachbarte Gebiete, wie Hinterkopf, Schultern und Schulterblätter oder auch thorakal, ein (Moorahrend, 1995).
Kopfschmerz
Kopfschmerzen nach einem HWS-Trauma setzen durchschnittlich etwa 5 Stunden nach Beginn des Traumas ein und es ergibt sich eine mittlere posttraumatische Kopfschmerzdauer von 21 Tagen (Keidel, 1995). Es kann auch zu diskontinuierlicher Kopfschmerzrückbildung kommen.
Armschmerzen
Bei etwa 20 % der Patienten zieht der Nackenschmerz bis in die Oberarme, und es kann ein brachialer Schmerz bis in die Unterarme und die Finger hinzukommen. Die Armschmerzen treten auch häufig bei ansonsten unauffälligen neurologischem Status auf.
Gefühlstörungen und motorische Beschwerden
Das Gefühl des Eingeschlafenseins, kribbelnde Mißempfindungen in den Händen und Unterarmen sowie das Gefühl verminderter Kraft in den Armen liegt zwischen 25 % und 56 % (Keidel, 1995).
Sehstörungen, Hörstörungen, Tinnitus und Gleichgewichtsstörungen (Hülse, 1997)
Zentrale Defizte treten auf. Es sollen sich bis zu 70 % auffällige EEG-Befunde und 62,5 % pathologische. Elektronystagmographiebefunde in der Literatur finden lassen (Thoden, 1993).
Vegetative Beschwerden
Etwa 70 % der Patienten klagen über Übelkeit, orthostatische Dysregulation, Kältegefühle, Schweißausbrüche und Neigung zu Hyperhidrosis.
Beschwerdefreies Intervall
Das Nackensyndrom und Muskelhartspann, Einschränkung der Kopfbewegungen und Berührungsempfindlichkeit im Nacken entwickelt sich in einem Intervall von 4 – 16 Stunden nach dem Trauma mit einem Gipfel bei 12 Stunden (Erdmann, 1973). Dieses beschwerdefreie Intervall tritt bei etwa 30 – 37 % der Verletzten auf (Wiesner und Mumenthaler, 1975) Wiesner und Mumenthaler sahen bei 10 % einen beschwerdefreien Intervall bis zu 48 Stunden. Dabei bestehen Beziehungen zum Schweregrad des Schleudertraumas. Graduiert man nach Erdmann, so bestehen beim schwersten Grad (Grad III) sofort Symptome, d.h. kein beschwerdefreier Intervall. Beim Grad II kommt es nach 4 – 8 Stunden und beim Grad I etwa nach 12 – 16 Stunden zu zunehmenden Beschwerden. Nach Erdmann kann das Beschwerdemaximum auch erst nach 14 Tagen erreicht werden.
Schweregrade
Der Einstufung nach Schweregraden liegt häufig das Schema nach Erdmann (1973) zu Grunde. Modifikationen der Schweregradeinteilung sind häufig vorgenommen worden und unterliegen auch heute noch der Kritik. Die brauchbarste Graduierung scheint nach wie vor die Einstufung von Erdmann zu sein (Schröter, 1995).
Rückbildung und Symptomfreiheit
Die Rückbildung geschieht innerhalb von Tagen und Wochen. In einer Studie von Deans et al. (1987) sind etwa 50 % der Patienten mit Nackenbeschwerden nach einem Vierteljahr beschwerdefrei. Die Rückbildung bis zur Symptomfreiheit kann langsam vorangehen. So können sich Beschwerden schon innerhalb von 2 Monaten zurückbilden, Symptomfreiheit tritt aber erst nach 2 Jahren auf (Maimaris et al., 1988).
Prolongierte bzw. chronische Verläufe
Hohl (1974) berichtet über bis zu 5 Jahre dauernde posttraumatische Beschwerdepersistenz mit einer mittleren Beschwerdedauer bei 2 Jahren (146 Patienten). Bei Gutachtenpatienten fanden Wiesner und Mumenthaler auch nach 3 – 6 Jahren intermittierende Restbeschwerden, wie Beeinträchtigung der Kopfbeweglichkeit oder Dolenz der paravertebralen HWS-Muskulatur. Bei Radanov et al. (1991) zeigten Patienten mit chronischem Verlauf (länger als 6 Monate) 95 % Nackenschmerzen, 71 % Kopfschmerzen, 88 % Müdigkeit, 65% Schlafstörungen, 57 % Schulterschmerzen.
Ursachen für protrahierte Rückbildung bzw. Chronifizierung
Pathologische posttraumatische Röntgenbefunde (Friedburg u. Nagelmüller, 1997; Huguenin et al., 1997)mit Subluxation oder Fraktur, vorbestehende degenerative HWS- Veränderungen verschlechtern die Prognose. Auch eine posttraumatische Umkehr der zervikalen Lordose und verminderte Beweglichkeit in einem HWS-Segment, die Erfordernis einer Halskrause für länger als ein Vierteljahr oder die Wiederaufnahme physikalischer Therapie nach zwischenzeitlicher Beschwerdefreiheit (Hohl, 1974) sind Faktoren eines protengierten Verlaufs.
Neurologische Defizite (Schmerzen, Reiz- und Ausfallerscheinungen) an Händen und Armen lassen eher eine ungünstige Prognose erwarten (Maimaris et al., 1988; Hohl, 1989). Kommt es neben radikulären Beschwerden zu gleichzeitigen muskulären Schwächen, so ergibt sich ein signifikanter Unterschied zwischen berenteten und den sogenannten Schadenfällen (Dvorak und Sandler, 1995).
Das Vorhandensein von initial ausgeprägten Kopf- und Nackenschmerzen, pseudoneurasthenischen Symptomen, depressiver Verstimmung, Befindlichkeitsstörungen und eine Einschränkung der passiven HWS-Beweglichkeit bei Inklination, treten bei verzögertem Beschwerderückgang auf.
Lampe (1995) fand bei 60 Verunfallten mit posttraumatisch chronischen Beschwerden segmentale Lockerungen im oberen und mittleren HWS-Bereich bei 81.7 %.
Psychische Unfallfolgen und Chronizität
Smith, 1989 und Radanov et al., 1989 finden Konzentrationsstörungen (65 %), depressive Stimmungslage (51 %), Schlafstörungen (51 %), Müdigkeit (83 %) sowie andere psychische Symptome, wie Benommenheit, Alpträume, sich aufdrängende Erinnerungen und Stressintoleranz. Dieser Beschwerdekomplex wurde immer wieder mit verschiedenen diagnostischen Begriffen gefaßt, wie z.B. traumatische Neurose, Kompensationsneurose, Anpassungsstörung, Belastungs-reaktion, psychogenes Schmerzsyndrom. Eine sichere systematische Gliederung gelingt nur schwer (Kallert, 1995).
Es kann auch bei psychischer Fehlentwicklung zu einer Dekompensation durch den Unfall kommen, so insbesondere zu ängstlichem Erleben, depressivem Erleben, Fehlentwicklung zu Konversionsreaktionen und Fehlentwicklung zu narzißtisch depressivem Erleben. Als Symptome würden sich dann bei ängstlichem Erleben starke Ängste und Phobien, bei depressivem Erleben intensive depressive Symptomatik, psychosomatisch psychovegetative Beschwerdekomplexe und bei Konversionsreaktion funktionelle Lähmungen und Defizite sensorischer Funktionen ergeben. Chronische Schmerzsyndrome bei einer Fehlentwicklung zu narzißtisch depressivem Erleben (Dahlmann, 1992).
Eine akute Belastungsreaktion bei Verkehrsunfallopfern setzt häufig direkt nach dem Unfall ein und tritt mit vegetativen Zeichen auf. Auch kommt es zu Angst, Depression, Ärger, Verzweiflung, Überaktivität und Rückzug. Die Rückbildung läuft innerhalb von wenigen Tagen ab.
Hiervon anzugrenzen ist die posttraumatische Belastungsstörung, bei der es durch das traumatisch Erlebnis zu Schlafstörungen, Reizbarkeit, Konzentrationsstörung, Hypervigilanz, Schreckreaktionen kommt. Das Ereignis muß außerhalb der üblichen menschlichen Erfahrung erlebt sein und Erinnerungen, Träume, Handeln und Fühlen so beeinflussen, als ob das traumatische Ereignis wiedergekehrt wäre.
Therapeutische Grundsätze
Prospektive Studien mit randomisierten Gruppen gibt es nicht genügend, um hier vollkommene Klarheit zu erlangen. In einer Studie von Maely et al. kommt es zu einem signifikanten Unterschied bei Schmerz und Beweglichkeit zu Gunsten einer frühen Mobilisation/Manipulation verglichen mit einer Ruhe-Wärme-Applikation. Häufig wird in der Initialphase eine Ruhigstellung mittels weichem oder hartem Halskragen empfohlen bei gleichzeitig analgetischer und muskelrelaxierender medikamentöser Behandlung. Nach 4-6 Wochen ist es angebracht, eine mobilisierende Behandlung einzuleiten, wobei neuromuskuläre Therapien zum Zuge kommen sollen (Schneider W., Dvorak, Dvorak-Tritschler, 1989). Bei Verletzungen mit starken muskulären Verspannungen sowie Triggerpunkten können Lokalinfiltrationen mit einem Anästhetikum zusammen mit Kälteapplikationen zur Anwendung kommen. Massagen mit Wärmeapplikation sind beliebt, bringen jedoch nur selten entscheidenden Durchbruch. Häufig steht auch die Behandlung der verkürzten tonischen Muskulatur im Vordergrund (Moorahrend, 1995). Es bedarf einer kompetenten Analgesie, einer kurzfristigen aber intermittierenden Ruhigstellung in der Schanz’schen Krawatte und einer frühzeitigen Aufnahme aktiver schneller Kopfbewegungen. Dehnungen mit Provozieren von Schmerzen (notizeptive Afferenzen) sollen vermieden werden (Lohse-Busch et al., 1997).
Im Patientenumgang sollte dem Verunfallten eine angemessene Information über das Distorsionstrauma sowie klare Verhaltensrichtlinien mit kurzen Kontrolluntersuchungen angeboten werden (Kügelgen, 1995). Er hält die intermittierende Versorgung mit der Schanz’schen Halskrawatte für völlig unsinnig, dies führe zu neuen Beschwerden infolge muskulärer Insuffizienz und Problemen der Krankheitsbewältigung. Die Patienten mit einer leichten HWS-Distorsion und anhaltenden Beschwerden haben mit hoher Wahrscheinlichkeit einen Therapieschaden (Kügelgen, 1995). Eine optimale konsequente Betreuung des Kranken gerade in der wichtigen Zeit unmittelbar nach der Verletzung soll gewährleistet sein. Dazu zählen ausführliche Information des Kranken unmittelbar nach der Diagnosestellung über die Krankheit. Bestehen nur Beschwerden und kein objektiver Befund, so ist eine Fixierung der Halswirbelsäule nicht erforderlich. Liegt ein pathologischer Befund des Bewegungsapparates vor, ist eine zusätzliche Ruhigstellung zu erwägen. Der Kranke muß informiert werden, daß sich durch die Ruhigstellung eine muskuläre Dysbalance entwickeln kann. Ist nach einem Monat der Therapieerfolg nicht eingetreten oder nicht kurzfristig abzusehen, sollte eine Verletzung der Kopfgelenke von manualtherapeutischer Seite untersucht werden (Kügelgen, 1995).
Lymphdrainage bei Halswirbelsäulen-Distorsions-Trauma
Literaturangaben bei Lymphdrainage bei Halswirbelverletzungen gibt es wenig.
Bei 5 Patienten mit HWS-Distorsions-Trauma (3 akut, 3 chronisch) wurden nur Lymphdrainagen des Kopfes, Nackens und der Schulter zweimal pro Woche für eine Dauer von etwa 30 Minuten durchgeführt. Bei den akuten Patienten kam es bereits im Laufe von 6 Behandlungen zu einer deutlichen Verbesserung des Nackenschmerzes mit ausstrahlenden Beschwerden in die Arme und des Schwindels. Eine Patientin berichtete, daß der Effekt der Behandlung erst nach 3 – 4 Stunden nach der Behandlung mit einer deutlich besseren Kopfbeweglichkeit eingesetzt hätte und mit zunehmenden Behandlungsanwendungen jeweils früher und länger anhaltend zu spüren gewesen sei.
Bei den chronischen Patienten, die vorhergehend nur unwesentlich von Massagen und anderen physikalischen Behandlungsmethoden profitiert hatten, nahm die Auslösehäufigkeit von Schwindel und Nackenschmerzen subjektiv deutlich ab.
Lymphdrainage als einzig durchzuführende Methode nach akuten und bei chronischen HWS-Distorsions-Traumata scheint einen direkt heilenden Effekt auf die Beschwerden zu haben und sollte in kontrollierten Studien untersucht werden.
Zusammenfassung
Beim Halswirbelsäulen-Distorsions-Trauma finden sich typische Beschwerdebilder mit guter Rückbildungstendenz. Bei bis zu einem Drittel der Patienten kommt es zu chronischen Beschwerden. Diese treten insbesondere bei psychischer Begleitsymptomatik und bei Kombination von radikulären Defiziten und Muskelschwächen auf. Studien mit eindeutiger Aussage zum optimalen therapeutischen Vorgehen gibt es wenige, reproduzierte Studien gibt es nicht. Fallbeispiele zeigen, daß die Lymphdrainage des Kopf-Nacken-Schulter-Bereichs bei akuten und chronischen Verläufen einen hohen therapeutischen Effekt hatte.
Autor: Prof. Dr. med. Klaus Peter Westphal, 1998
Fragen zu Behinderungsgrad, Behördliche Auseinandersetzung, Gutachten: www.global-help.de
Verteilung der Erkrankung in der Bevölkerung (Epidemiologie) (Update 2020)
Demenzerkrankungen zeichnen sich durch den Abbau und Verlust kognitiver
Funktionen und Alltagskompetenzen aus. Der Verlauf ist in der Regel progressiv und es
kommt u.a. zu Beeinträchtigungen der zeitlichen und örtlichen Orientierung, der Fähigkeit
zu Kommunizieren, der autobiographischen Identität sowie zum Verlust von
Persönlichkeitsmerkmalen. Das schwere Stadium der Demenz ist häufig durch
vollständige Hilflosigkeit und Abhängigkeit von der Umwelt gezeichnet. Demenzpatienten
haben ein erhöhtes Morbiditätsrisiko für andere Erkrankungen und eine verkürzte
Lebenserwartung.
Für Angehörige entsteht ebenso wie für den Patienten eine hohe emotionale Belastung.
Pflegende Angehörige von Demenzerkrankten haben ein erhöhtes Risiko für psychische
und körperliche Erkrankungen.
Aufgrund der demographischen Entwicklung sind Demenzerkrankungen ein zunehmendes
gesellschaftliches Problem. Aktuell leben etwa 1,6 Mio. Demenzerkrankte in Deutschland,
eine Verdoppelung bis 2033 ist zu erwarten.
Derzeit werden jährlich 5,6 Milliarden Euro von den gesetzlichen Krankenversicherungen
für Demenzerkrankte ausgegeben (Statistisches Bundesamt (Hrsg): Gesundheit.
Ausgaben, Krankheitskosten und Personal 2004. Wiesbaden, Statistisches Bundesamt
2006).
© 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Ätiologie (Update 2020)
Es werden verschiedene Formen der Demenzerkrankung unterschieden:
- Typische und atypische Alzheimererkrankung, mit 45-70% die häufigste Form der
Demenz - Vaskuläre Demenz (15-25%)
- Gemischte Demenz (5-20%)
- Frontotemporale Demenz/Morbus Pick (3-18%)
- Lewy-Körperchen-Demenz (3-10%, assoziiert mit M. Alzheimer und Parkinson, genaue
Ätiologie unklar)
Neue Kriterien der Alzheimererkrankung – IWG-2-Kriterien für die Alzheimer-
Krankheit (Dubois et al., 2014)
Typische Alzheimer-Krankheit
- Episodische Gedächtnisstörung, isoliert oder in Verbindung mit Beeinträchtigungen
in anderen kognitiven oder Verhaltensdomänen, die langsam fortschreitet und
mindestens 6 Monate besteht und von dem Patienten oder einem Informanten
beschrieben wird. Die Gedächtnisstörung ist vom hippokampalen Typ, dokumentiert
durch reduzierte Leistung in einem Test der episodischen Gedächtnisleistung, der
für die Hippokampusfunktionen spezifisch ist, z.B. cued recall mit kontrollierter
Enkodierungsphase. - Hinweise für die Pathologie der Alzheimer-Krankheit durch mindestens eines der
folgenden Kriterien:- Erniedrigtes Aß42 im Liquor und erhöhtes Tau-Protein bzw. phosphoryliertes
Tau-Protein im Liquor - Positiver Amyloid-Nachweis mit PET
- Mutation, die zu einer monogen vermittelten Alzheimer-Krankheit führt
(Mutation auf den Genen Presenilin 1 oder Presenilin 2 oder auf dem Gen des
Amyloid-Precursor-Proteins, APP)
- Erniedrigtes Aß42 im Liquor und erhöhtes Tau-Protein bzw. phosphoryliertes
Ausschlusskriterien:
- Plötzlicher Beginn der Symptomatik
- Frühe Gangstörungen, frühe Krampfanfälle, schwere frühe
Verhaltensänderungen, frühe extrapyramidalmotorische Zeichen oder Halluzinationen - Fokaleneurologische Zeichen
- Fluktuation der kognitiven Störungen
- Andere Erkrankungen, die die kognitiven Störungen erklären können
Atypische Alzheimer-Krankheit
Junge Alzheimerpatienten zeigen häufig atypische Varianten, z.B. mit Schwerpunkt auf
einer Verarbeitungsstörung visueller Information (posteriore kortikale Atrophie),
Verhaltensstörungen oder Sprachstörungen (frontale oder logopenische Variante).
Für weitere Information/Quelle:
Leitlinie DGN „Demenzen“Copyright
© 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Differentialdiagnostik
Neuere wissenschaftliche Meinungen gehen davon aus, dass es eher wahrscheinlich ist, dass die verschiedenen Demenz-Syndrome durch so genannte pathogenetische Kaskaden verursacht werden, diese pathogenetischen Kaskaden sich aber gegenseitig beeinflussen und unterschiedlich überlagern. Insgesamt ist es so, dass auch nach klinischen Kriterien eine Demenzerkrankung nicht immer klar abgrenzbar gegen eine andere Demenzerkrankung ist. Andererseits können die klassischen Demenzerkrankungen in ihrem klinischen Verlauf und in ihren Ausfallerscheinungen und auch im Verteilungsmuster ihrer neuropathologischen Auffälligkeiten häufig gut voneinander getrennt werden. Aus pragmatischen Gründen werden deshalb die neurodegenerativen Erkrankungen getrennt. Die Diagnosestellung in neurologischen und nervenärztlichen Praxen versucht nach diesen Unterteilungen vorzugehen.
Diagnose des Demenz-Syndroms
Es werden die Beschwerden vom Patienten und insbesondere von Angehörigen geschildert (Fremdanamnese) und es werden orientierende neuropsychologische Tests durchgeführt, die insbesondere die geistige Leistungsfähigkeit des Betroffenen einschätzen lassen und dem Arzt die Sicherheit geben, dass überhaupt verminderte geistige Leistungen vorliegen.
Der Abbau von Hirngewebe bei den Demenzen lässt sich durch computertomographische oder kernspintomographische Untersuchungen dokumentieren und es lassen sich auch auf Grund dieser Bilder diagnostische Einschätzungen ableiten. Beispielsweise treten bei Demenzen vaskulärer Genese andere Auffälligkeiten in den so genannten bildgebenden Methoden auf als bei den primären degenerativen Formen. Auch die Verteilung der Hirnatrophie auf bestimmte Hirnregionen lässt diagnostische Schlüsse zu. Zudem können mit der bildgebenden Diagnostik auch andere Erkrankungen, die so genannte sekundäre Demenzformen verursachen, erkannt werden. Ein Normaldruck-Hydrocephalus kann ausgeschlossen werden, ein Subduralhaematom sowie auch seltene entzündliche Erkrankungen des Gehirns oder auch Hirntumore, die zu geistigem Abbau hätten führen können.
Der Hirnabbau bestimmter Hirnstrukuren führt auch zu einer Veränderung der Hirnströme (Elektroenzephalogramm, EEG). Die Oszillationen im EEG bzw. der Rhythmus der Potentialschwankungen der Hirnströme verlangsamt sich häufig. Es finden sich auch bei aktivierenden Aufgaben für die Hirnfunktionen nicht die typischen Veränderungen im EEG, wie man es bei Gesunden erwarten würde.
Untersuchungen des Nervenwassers (Liquor) können ebenfalls wichtig sein, um seltene entzündliche Erkrankungen als Ursache der Demenz auszuschließen.
Nuklearmedizinische Bilder des Gehirns
Nuklearmedizinische Bilder des Gehirns, die anzeigen, wie der Stoffwechsel der Hirnzellen in den jeweiligen Hirnarealen funktioniert, geben ebenfalls Hinweise auf Demenzerkrankungen. Hier wird die Methode der Positronen-Emissions-Tomographie (PET) und die Methode der Single-Photon-Emissions-Tomographie (SPECT) angewendet.
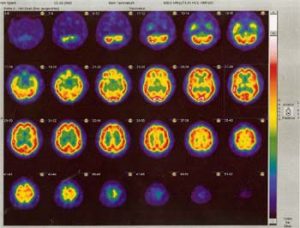
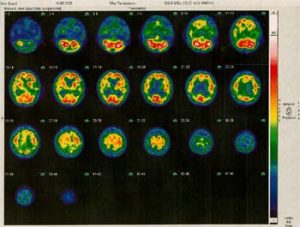
Die Aufgabe des Neurologen, Nervenarztes oder Psychiaters ist es also, zunächst eine Demenz überhaupt zu erkennen und zu beweisen und zum anderen andere Erkrankungen, die ähnliche Symptome verursachen könnten, auszuschließen bzw. ebenfalls zu erkennen.
Das cholinerge System und EEG-Veränderungen bei Demenz
Veränderungen der EEG-Aktivität unterliegen verschiedenen Transmitter-Systemen. Deutlich wirkt das cholinerge System auf die EEG-Modulation. Eine fronto-basale Kerngruppe von Neuronen im Gehirn (Nucleus Basalis Meynert) schickt überwiegend Nervenfasern zu anderen Nervenzellen, die zu diesen Nerven mit Hilfe des Überträgerstoffs Acethylcholin funktionieren. Diese so genannten cholinergen Neuronen degenerieren insbesondere bei der Alzheimer-Erkrankung. Dies führt zur Verlangsamung des EEG und auch zur verminderten Ansprechbarkeit des EEG-Rhythmus auf Sinnesreize oder andere Reize und Anforderungen im Gehirn.
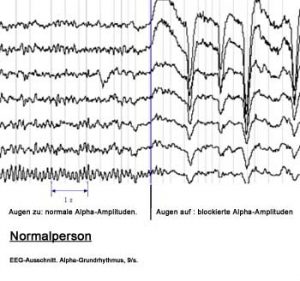
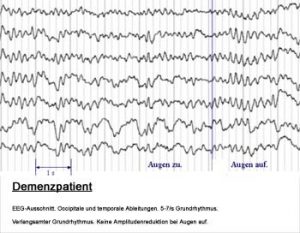


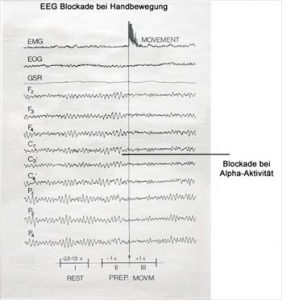
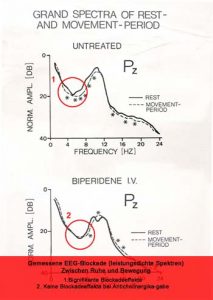
Beispiele
Beispielsweise führt eine Handbewegung zu einer Blockade einer Alphaaktivität. Gibt man bei gesunden Personen unter wissenschaftlichen Fragestellungen ein Medikament, das anticholinerg wirkt, also die Übertragungen der cholinergen Nervenzellen behindert, so vermindert sich die Aktivierungsfähigkeit des EEG bzw. die Blockademechanismen werden verringert. Dies ist im Anhang in Abbildungen ausgedrückt.
Auch bei der Alzheimer Demenz liegen ähnliche Effekte vor, wie man sie bei Gesunden unter Anticholinergikagabe erreicht wie in dem Beispiel (Bild) dargestellt. Es kommt bei Alzheimer-Patienten nicht zur Unterdrückung der Alphaaktivität bei Augenöffnen während einer EEG-Ableitung, sondern die Alphaaktivität bleibt unbeeinflusst. Dies kann gemessen und auch bildlich dargestellt werden. Alzheimer-Patienten zeigen in einer topographischen farbigen Darstellung (Mapping) verlangsamte Alpha- bzw. Grundaktivität und verminderte Amplitudenreduktion, wenn sie die Augen öffnen. Dies kann auch zusätzlich in der klinischen Diagnose verwendet werden.
Therapie der Alzheimer-Erkrankung (Update 2020)
Den Defekt in der cholinergen Übertragung der Nervenzellen kann dadurch beeinflusst werden, dass das Enzym, welches das Acethylcholin abbaut, behindert wird. Diese Substanzen werden Cholinesterasehemmer genannt. Dazu zählen
- Donepezil
- Rivastigmin
- Galantamin.
Auch wirkt bei Alzheimer-Erkrankung Memantine, das das glutamaterge System verbessert.
Sämtliche Medikamente dürfen nur vom Arzt verordnet werden und unterliegen ständigen Kontrollkriterien. Sie sind rezept- und apothekenpflichtig. Diese Substanzen dürfen auf der Grundlage der hier gegebenen Informationen nicht vom Leser oder durch weitergegebene Informationen gekauft oder eingenommen werden. Der Autor übernimmt keinerlei Verantwortung für die Nennung der therapeutisch wirksamen Medikamente in der Homepage.
In der Forschung finden sich weitere Studien zur Überprüfung von Impfstrategien gegen Alzheimer (amyloidreduzierende Methoden) . Auch wirken nicht-steroidale Antiphlogistika gegen die Alzheimer-Erkrankung. Antioxydative, hormonelle und lipidsenkende Ansätze werden seit langem verfolgt. Ähnlich wie bei der Parkinson-Erkrankung gibt es Versuche mit Stammzellen zur Regeneration untergegangener Neurone bzw. ihrer Transmissionen.
Pathologie der Alzheimer Demenz
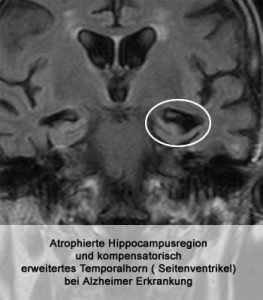
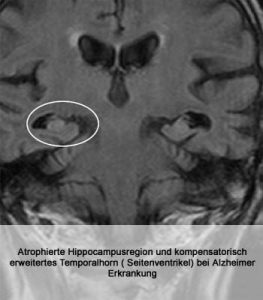
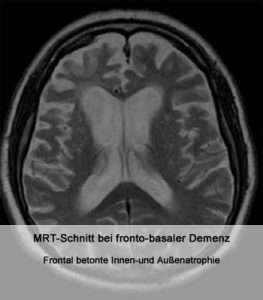
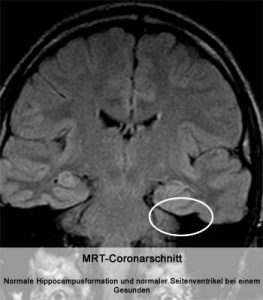
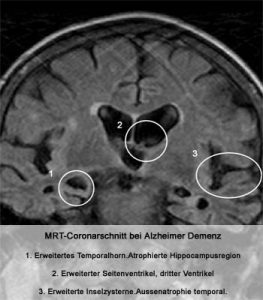
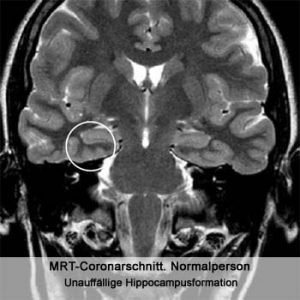
Die Abbildungen zeigen, ähnlich wie auch die dargestellten computertomographischen und kernpsintomographischen Bilder „Querschnitte durch das Gehirn“ Verschmächtigung des Gehirns in allen Anteilen. Mikroskopisch lassen sich entsprechend typische Veränderungen finden, wie sie auch auf den Bildern beschrieben sind, mit Fibrillen, Plaques und Lewy-Körperchen (Darstellung mehrerer Bilder).

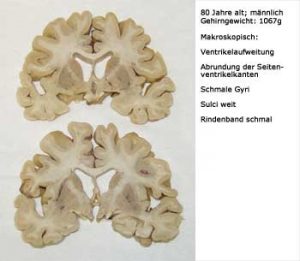

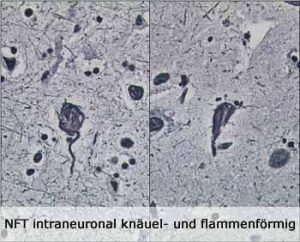
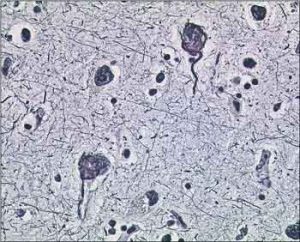
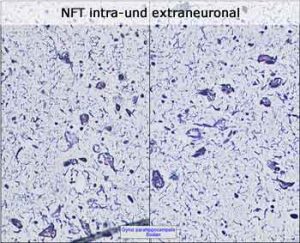


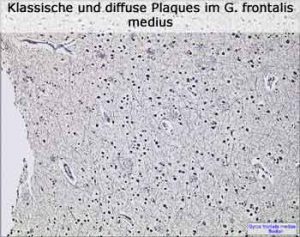
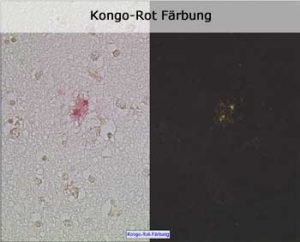
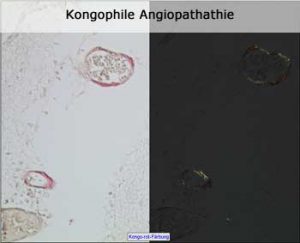



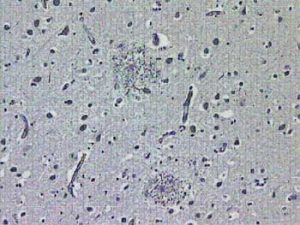

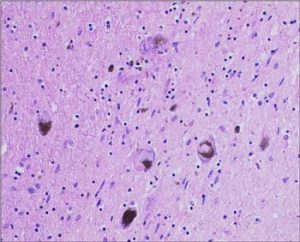
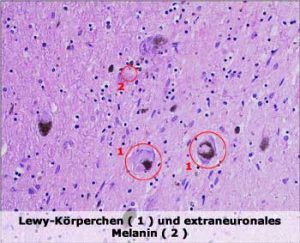
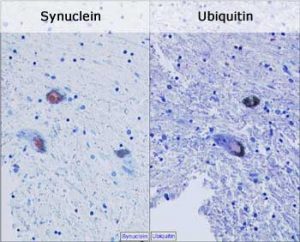
Methoden der Demenz-Diagnostik
Bei Gedächtnisstörungen werden Tests zur Früherkennung von Demenz durchgeführt, u. a. DemTec. Untersucht wird das EEG mit Mapping Methoden, CCT (Computertomographie des Kopfes), Kontrolle der Hirndurchblutung (Duplex – Sonographie der Gefäße). Alle Methoden dienen der Diagnostik von Gedächtnisstörungen und Demenz. Der Schweregrad von Demenz lässt sich bestimmen, auch der Grad von Arterioskelerose. Wichtige andere Erkrankungen des Gehirns, die auch Gedächtnisstörungen hervorrufen, können erkannt werden.
Frau Prof. Dr. Dr. Angelika Scheuerle (Abt. Neuropathologie der Prosektur am Bezirkskrankenhaus Günzburg) danke ich für die Überlassung Ihrer anatomischen und pathologischen Beispiele.
angelika.scheuerle@bkh-guenzburg.de
Hr. Dr. med. Markus Palmbach (Radiologie/Neuroradiologie) und Hr. Dr. med. Dieter Wanjura (Nuklearmedizin) danke ich für die Überlassung der Kernspinbilder und der SPECT-Bilder.
www.radiologiezentrum-ulm.de
Therapie der Alzheimer-Erkrankung - Ausblick (Update 2020)
Die bisher zur Verfügung stehenden Medikamente zur Behandlung einer Alzheimer-
Erkrankung zielen auf eine Verbesserung des Mangels an Neurotransmittern
(Botenstoffen) ab. Besonders Acetylcholin spielt hierbei eine wichtige Rolle, entsprechend
werden Acetylcholinesteraseinhibitoren eingesetzt (siehe oben). Auch NMDAAntagonisten
wie Memantin werden verwendet.
Einen neuen Ansatz verfolgt der monoklonale Antikörper Aducanumab, der lösliche
Oligomere und Ablagerungen von Beta-Amyloid, wie sie bei der Alzheimer-Erkrankung
vorkommen, binden soll. Dadurch soll es zu einer Reduktion des Amyloidlevels kommen.
Es wäre der erste ursächliche Therapieansatz. In tierexperimentellen Studien war ein
Fortschreiten der neurodegenerativen Entwicklung durch den Antikörper verhindert
worden.
Erste klinische Studien an Patienten hatten zunächst ernüchternde Ergebnisse erbracht
(EMERGE und ENGAGE-Studie), eine kürzlich erfolge neue Auswertung der Daten hatte
jedoch laut Herstellerkonzern Biogen gezeigt, dass der Antikörper in höherer Dosierung
doch in der Lage sei, den kognitiven Abbau bei Alzheimerpatienten im Frühstadium zu
verringern. Im Vergleich zu Placebo kam es unter Aducanumab 78 Wochen nach
Therapiebeginn zu einer signifikanten Reduktion der klinischen Verschlechterung
gegenüber dem Ausgangswert (EMERGE 2019).
Der Konzern Biogen plant, für Aducanumab 2020 die Zulassung bei der amerikanischen
Arzneimittelbehörde FDA zu beantragen. Das wäre der erste Schritt, um den Wirkstoff als
Medikament verfügbar zu machen.
Für weitere Information/Quelle:
Leitlinie DGN „Demenzen“
aerzteblatt.de „Alzheimer-Wirkstoff Aducanumab“
Alzheimer Forschung Initiative e.V.
Holger Grehl, Frank Reinhardt: Checkliste Neurologie, Georg Thieme Verlag, Stuttgart
2016
A. Hufschmidt, C.H. Lücking, S. Rauer: Neurologie compact, Georg Thieme Verlag,
Stuttgart 2013
Copyright © 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Multiple Sklerose
Die Multiple Sklerose (MS, syn. Encephalomyelitis disseminata) ist eine chronische,
autoimmun vermittelte multifokale Erkrankung des zentralen Nervensystems (ZNS). Aus
bisher unbekannten Gründen kommt es zu punktuellen Entzündungen der
Myelinscheiden (die elektrisch isolierende äußere Schicht der Nervenfasern) im ZNS.
Das Myelin im peripheren Nervensystem ist klinisch nicht relevant betroffen. Somit können
Entzündungsherde im Gehirn, Rückenmark und in zentral myelinisierten Hirnnerven (N.
opticus) auftreten. Die Entzündung führt zu fokalen Demyelinisierungen, im Verlauf treten
gliöse Vernarbungen auf.
Epidemiologie: Prävalenz ca 30-80/100 000 Einwohner in Europa, jährliche Inzidenz ca
4-6/100 000 Einwohner.
Risikofaktoren: Verwandte 1. Grades haben ein ca 20fach erhöhtes Erkrankungsrisiko,
Rauchen, Vit. D-Mangel und bestimmte Infektionen (EBV, HHV6) sind ebenfalls mit einem
erhöhten Risiko vergesellschaftet.
Das mittlere Erkrankungsalter beträgt 28 Jahre. MS ist nicht heilbar, der Verlauf der
Erkrankung kann in den meisten Fällen jedoch sehr positiv beeinflusst werden. Nicht
zwangsläufig kommt es zu schwereren Behinderungen. 25 Jahre nach Erstdiagnose sind
1/3 der Pat. noch arbeitsfähig, 2/3 noch gehfähig.
Die Prognose ist enorm abhängig von der Verlaufsform der MS. Man unterscheidet:
- Klinisch isoliertes Syndrom (CIS): Erste typische neurologische Funktionsstörung.
Eine zeitliche Dissemination (s.u.) ist nicht nachweisbar. Entwicklung einer MS möglich. - Schubförmig remittierende Verlaufsform (RRMS): ca 85-90%, es kommt hierbei zu
einzelnen Erkrankungsschüben mit folgender vollständiger oder inkompletter
Rückbildung der Symptomatik, zwischen den Schüben kann weitgehende oder
komplette Beschwerdefreiheit bestehen. - Sekundär progrediente Verlaufsform (SPMS): ca 40-50% Übergang von RRMS zu
SPMS, mit oder ohne aufgelagerte Schübe, aber mit kontinuierlicher klinischer
Verschlechterung. - Primär chronisch progrediente Verlaufsform (PPMS): ca 10%, schleichende
Zunahme der Funktionsstörungen ohne Schübe.
Prognostisch günstig sind: schubförmige Verlaufsform, monosymptomatische Schübe (nur
ein Symptom), gute und lang anhaltende Remission nach dem ersten Schub, Beginn vor
dem 40. Lebensjahr, kernspintomografisch geringe Herdlast (v.a. in der Frühphase).
Klinische Befunde
Entsprechend der Lokalisation der Herde im ZNS können Defizite aller Hirnfunktionen
auftretend. Die Symptome nehmen im Schub meist innerhalb einiger Stunden oder
weniger Tage zu und bilden sich spontan langsam – teilweise oder vollständig – zurück.
Häufig sind:
- Okuläre Symptome: Visusverlust/Schleiersehen, typisch für eine Retrobulbärneuritis
(Entzündung des Sehnervs, häufigste Erstmanifestation bei jungen Patienten). Auch
Doppelbilder durch Paresen der Augenmuskeln können auftreten. - Sensibilitätsstörungen jeglicher Art (Taubheitsgefühle, Kribbelparästhesien, etc)
- Motorische Störungen: Spastische Paresen u.U. mit Gangstörung, Reflexsteigerung
- Zerebelläre Symptome (vom Kleinhirn ausgehend): Ataxie (Koordinationsstörung),
Sprachstörung, Nystagmus (Augenbewegungsstörung) - Autonome Störungen: v.a. Blasen- und Sexualfunktionsstörung
- Psychosyndrom: euphorische oder depressive Stimmung
- körperliche und geistige Erschöpfbarkeit (Fatigue)
Diagnostik
Die Diagnose einer MS kann nur gestellt werden, wenn die Kriterien der räumlichen und
zeitlichen Dissemination erfüllt sind. Das bedeutet, dass der Nachweis einer 1)
entzündlichen, 2) multifokalen und 3) mehrzeitigen Erkrankung des ZNS gegeben sein
muss.
Die Diagnostik beinhaltet also neben einer körperlichen Untersuchung und der Anamnese
in erster Linie eine Nervenwasseruntersuchung (Nachweis eines chronisch
entzündlichen Zellbildes sowie spezifischer Antikörper) und eine Bildgebung mittels MRT.
Im MRT kann der Nachweis verschiedener Entzündungsherde gelingen, wenn diese die
Kriterien der räumlichen und zeitlichen Dissemination erfüllen und eine passende Klinik
besteht, kann von einer MS gesprochen werden.
In vielen Fällen ist die Diagnostik aber nicht eindeutig, sodass besonders nach einem
ersten schubähnlichen Ereignis häufig (noch) nicht die Diagnose MS gestellt werden kann.
Differentialdiagnosen
Die Diagnose MS ist nur möglich, wenn die neurologische Symptomatik durch nichts
anderes besser erklärt werden kann. Differentialdiagnostisch kämen in Frage:
- Immunvaskulitiden und ZNS-Beteiligung systemischer Immunerkrankungen (z.B. Lupus
erythematodes, rheumatoide Arthritis, Sarkoidose, etc) - Neuromyelitis optica
- Infektionen (Neuroborreliose, Neurosyphilis, etc)
- Multiple zerebrale Embolien
Therapie des akuten Schubes
Ein akuter Schub wird mit einer Kortisonstoßtherapie intravenös behandelt. Je nach
Schwere des Schubes und der Gesamtsituation ist dafür häufig eine stationäre
Behandlung notwendig. Die Dauer der Therapie beträgt meist 3-5 Tage. Darunter kommt
es i.d.R. zu einer raschen Rückbildung der Schubsyptomatik. Eine sichere Beeinflussung
des Langzeitverlaufs der Erkrankung ist nicht belegt. Deswegen bedarf es unabhängig von
der Therapie eines einzelnen Schubes einer Schubprophylaxe.
Intervalltherapie/Schubprophylaxe
Ziel der langfristigen Therapie ist es vor allem, weitere Schübe zu verhindern, den
Krankheitsprozess zu verlangsamen und damit der Entwicklung irreversibler
neurologischer Funktionsstörungen vorzubeugen. Deswegen sollte eine Therapie
möglichst früh begonnen werden, bereits beim Vorliegen eines CIS, spätestens nach der
Diagnosestellung einer schubförmigen MS-Verlaufsform.
Für eine frühzeitige Therapie bei CIS sprechen insbesondere:
- Klinisch signifikante Beeinträchtigung
- Nachweis oligoklonaler Banden (spez. Antikörper) im Nervenwasser
- Hohe Läsionslast (> 6 Herde) im MRT
- Schlechtes Ansprechen auf die Kortisonstoßtherapie
Bei der Wahl der Medikation zur Schubprophylaxe unterscheidet man zwischen milden/
moderaten und hochaktiven Verlaufsformen der schubförmigen MS. Als hochaktiv gilt
die Erkrankung, wenn trotz einer prophylaktischen Therapie weiterhin Schübe auftreten
und es zu einer Zunahme der Herdlast im MRT kommt. Bei Fehlen einer prophylaktischen
Therapie gilt die Erkrankung als hochaktiv, wenn es innerhalb 1 Jahres zu mindestens 2
Schüben mit Behinderungsprogression kommt und ebenfalls eine Zunahme der Herdlast
im MRT besteht.
Für die moderate Form der MS kommen therapeutisch u.a. Beta-Interferone,
Glatirameracetat, Teriflunomid und Dimethylfumarat in Frage. Ein relevanter Unterschied
bzgl. der Wirksamkeit besteht nicht, die Präparate unterscheiden sich primär durch ihre
Applikationsform (oral, subkutan), die Häufigkeit der Einnahme (täglich, wöchentlich) und
ihre potentiellen Nebenwirkungen.
Für die hochaktive Verlaufsform stehen medikamentös u.a. Fingolimod, Alemtuzumab,
Natalizumab und Cladribin zur Verfügung. Auch hier ist die Wirksamkeit der Medikamente
vergleichbar, die Wahl eines Medikaments erfolgt eher aufgrund möglicher
Nebenwirkungen.
Bei jeder langfristigen Therapie ist bei Frauen das Thema Schwangerschaft zu beachten
und ein Medikament muss dahingehend gewählt oder angepasst werden. Grundsätzlich
ist eine normal verlaufende Schwangerschaft bei MS-Patientinnen die Regel, tatsächlich
hat die Schwangerschaft häufig einen günstigen Einfluss auf den Krankheitsverlauf.
Disclaimer: Sämtliche Medikamente dürfen nur vom Arzt verordnet werden und unterliegen
ständigen Kontrollkriterien. Sie sind rezept- und apothekenpflichtig. Diese Substanzen
dürfen auf der Grundlage der hier gegebenen Informationen nicht vom Leser oder durch
weitergegebene Informationen gekauft oder eingenommen werden. Der Autor übernimmt
keinerlei Verantwortung für die Nennung der therapeutisch wirksamen Medikamente auf
der Homepage.
Für weitere Information/Quelle:
Leitlinie DGN – Multiple Sklerose (aktuell in Überarbeitung)
MSD Manual
Holger Grehl, Frank Reinhardt: Checkliste Neurologie, Georg Thieme Verlag, Stuttgart
2016
A. Hufschmidt, C.H. Lücking, S. Rauer: Neurologie compact, Georg Thieme Verlag,
Stuttgart 2013
Copyright © 2019 Lukas Mästle-Goer, Prof. Dr. med. Klaus Peter Westphal
Leistungen
Unsere Leistungsschwerpunkte
Erfahren Sie mehr zu unserer Leistungsangebot in den jeweiligen Themenbereichen.
Haben Sie Fragen
Sie möchten einen Termin vereinbaren?
Montag, Dienstag und Donnerstag:
07:00 – 12:00 Uhr und 14:00 – 17:00 Uhr
Mittwoch und Freitag:
07:00 – 12:00 Uhr
Wir freuen uns auf Ihren Kontakt.
Bitte bringen Sie zu den Untersuchungen alle wichtigen Unterlagen, Bilder, CDs, Befunde etc. mit.